| |
 |
Should you encounter any unexpected behaviour,
please let us know.
|
elNémo help !
- How do I submit a job to elNémo ?
A new job can be submitted using the link "start a new run".
Here are some things that you should know about.
- The input format is a protein structure in PDB format.
- Only ATOM records are used.
- Water molecules are filtered out (residues named WAT and HOH).
- Make sure that special residues like MSE and other atoms or molecules that are listed as HETATOM
in the PDB file, but that you wish to be used in the calculations (such as Calcium or Zink) are coded as ATOM.
- Single ions should be placed next to one of their fixing residues, so that they will be grouped
together into the same rigid block.
- Hydrogen atoms (sometime present in high resolution structures) and alternative conformations
should be deleted.
- The CUTOFF parameter defines which atom-atom interactions should be kept in the elastic network model.
If you wish to do computations using only C-alpha atoms, increase the CUTOFF to values around 12.
Otherwise the default value should be ok.
- The parameter NRBL defines how many residues should be treated as a rigid body. This parameter is
by default determined automatically as a function of protein size. NRBL automatically determines the computation time
of your run. Setting it to a low value for a large protein may cause your job to run for days (and eventually getting
killed by the system).
- By default 5 modes with 11 models for each mode will be computed. Animations (if requested) will be
drawn using Molscript. Typically this takes about 3 minutes per mode, but for
larger proteins (more 300 residues) this can become very time-consuming. Think about using a local
viewer such as VMD in in these cases.
You can always request additional mode computations and graphics once your job has finished.
- elNémo jobs will be executed one-at-a-time on a first-come first-served basis. The
"job status" page informs you about the status of your job (finished, spooled, running)
and the number of jobs that are waiting before yours.
- Computation of additional modes is done in a separate queue and in parallel.
- If you enter an e-mail address, your username will be used to identify your job in the queue
(your e-mail address will never be shown publicly and it will only be used to inform you
about the status of your job). If you do not wish to enter your e-mail address, you may still
enter a pseudonym (without a '@' character) to associate a username to your job.
- If you check the "keep my job private" checkbox, only your username will be visible to others.
In that case keep track of your job-id as it is the only means to access your job results.
- If you are encountering any problems with this server (i.e. hung jobs), do not hesitate to contact
me by

- What kind of output can I expect from elNémo ?
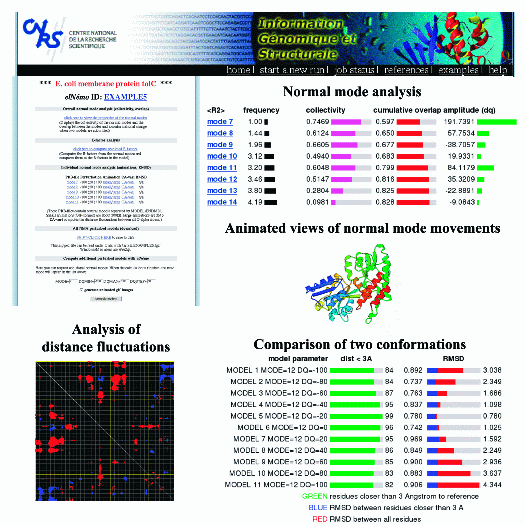
This is an example of a typical elNémo output that is available for every run through the result page (top left).
The normal mode analysis page (top right) displays the different properties of the first 100 lowest frequency modes,
that is their frequency, collectivity of atom movement, overlap of each mode with the observed conformational change
(if two conformations are available) and its corresponding amplitude.
3D animations from three orthogonal viewpoints are available in large and small sizes (produced with Molscript).
Comparison of a normal mode perturbed structure and a second conformations in terms of RMSD and number of residues that
are closer than 3A can be done (bottom right). Analysis of distance fluctuations between all CA atoms is presented in
the form of a cross-plot, where red and blue dots indicate those residues for which the distance changes significantly in movement
related to the given mode. The result page also allows to submit normal mode calculations for new modes or different
amplitudes, and to download the resulting models in PDB format for further processing (e.g. using VMD to visualize the
protein movements as presented on the elNémo example page).
- How can I make animations like those presented on the example page ?
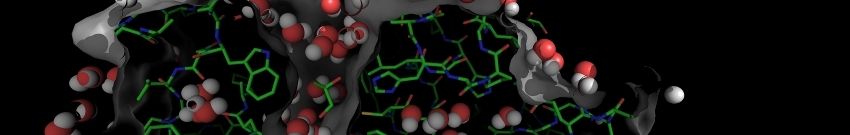
The images on the example page were all produced using
VMD and its internal Tachyon ray tracer.
Surface plots were made using MSMS via the VMD interface.
First, save the PDB file of a single mode from the result page of your run (e.g shift-click on mode 7
in the Individual normal mode analysis section of that page to save all conformations of mode 7).
Then load that PDB file into VMD. The animation should start automatically. You may want to adjust
the representation of the protein in VMD to van-der-waals or cartoon.
If you find results from this site helpful for your research, please cite one of our papers:
elNémo
is maintained by Yves-Henri Sanejouand.
It was developed
by Karsten Suhre.
Between 2003 and 2014, it was hosted by IGS (Marseille).
Last modification: April 25th, 2023.
|
|
|
|
|