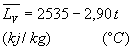Annexe 1 : Détermination expérimentale des caractéristiques thermodynamiques
Plan
1. Détermination expérimentale des coefficients
thermoélastiques et des équations détat
1.1. Cas des solides et des liquides
1.2. Cas des gaz
2. Calorimétrie
2.1. Mesure des capacités calorifiques
2.2. Chaleurs latentes de changement de phase
1. Détermination expérimentale des coefficients thermoélastiques, des équations détat
1.1. Cas des solides et des liquides
Nous ne décrirons pas les appareillages utilisables mais invitons le lecteur intéressé à se reporter à des ouvrages spécialisés.
Pour ce qui est de la connaissance du coefficient de dilatation à pression
constante des solides, on prend des solides en forme de fil et on mesure le
coefficient de dilatation linéaire à pression constante ![]()
Le coefficient de compressibilité isotherme sobtient à partir des modules de Young, Poisson et/ou de cisaillement mais ceci nécessite des connaissances sur les contraintes dans les solides.
Pour ce qui est de la connaissance du coefficient à pression
constante des liquides, la mesure se fait directement à partir de
lexpérience mise au point par Dulong et Petit ou à laide
dun dilatomètre à tige.
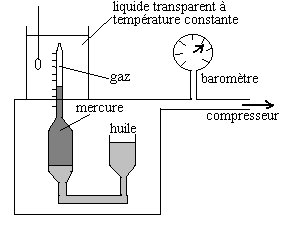
La mesure du coefficient de compressibilité isotherme pour les
liquides se fait directement dans des piézomètres à
capillaire.
Remarque : on ne mesure pas le coefficient b daugmentation de pression à volume constant parce quil est très difficile de faire subir à un liquide ou un solide une transformation isochore.
Equations détat des solides et des liquides
Pour les solides et les liquides, les coefficients a
et cT
sont faibles (en première approximation, on les considère
comme indilatables et incompressibles).
![]() est une équation
détat habituelle dans des limites de température et de pression
à préciser pour chaque corps.
est une équation
détat habituelle dans des limites de température et de pression
à préciser pour chaque corps.
Les liquides, qui font partie de létat fluide, obéissent,
aussi, à des équations détat de type Van der Waals
que nous verrons avec létude des gaz
 |
Les premières expériences de compressibilité
isotherme des gaz, dans des domaines de pression très limités
ne dépassant pas quelques atmosphères, ont été
faites au 17ème siècle par
Mariotte en France et par Boyle en Angleterre.
Plusieurs expérimentateurs (Regnault, Andrews, Cailletet et Amagat) ont repris ces études au 19ème siècle dans des domaines de pression de plus en plus élevés. Kamerlingh Onnes à Leyde a étendu les mesures au domaine des basses températures. |
Ces mesures ont été reprises au 20ème
siècle avec une précision accrue sur tous les gaz connus
dans des domaines de température extrêmes et pour des pressions
atteignant 1000 atmosphères.
De longues séries de mesures systématiques ont été
faites dans deux laboratoires spécialisés : en Europe celui
de Van Der Waals à Amsterdam sous la direction de Michels et aux
Etats -Unis au laboratoire du Massachusetts Institute of Technology sous
la direction de Beattie.
Nous présentons, à titre dexemple, une série
de résultats en coordonnées dAmagat p, pV pour le
diazote.
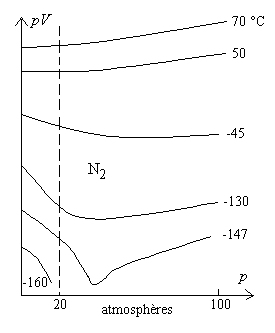 |
Au-dessous dune température (-147 °C
pour le diazote) les courbes sont interrompues car le gaz se liquéfie.
Cette température est appelée température critique
et notée TC .
Une autre température particulière apparaît, la température de Mariotte TM , où les courbes sont horizontales pour p = 0 ( Pour des températures supérieures à la température de Mariotte, les pentes pour p = 0 sont positives, elles sont négatives dans le cas contraire. Pour le gaz parfait, limite de comportement de tous les gaz réels lorsque la pression devient nulle, les courbes sont horizontales quelle que soit la température. Un gaz réel se rapproche au mieux du comportement du gaz parfait à sa température de Mariotte. |
Equations détat des gaz
Il nexiste pas déquation universelle rendant compte des courbes
ci-dessus.
Pour des valeurs faibles de p, nous devons retrouver léquation
détat des gaz parfaits pV=nRT
Des équations classiques sont donc :
![]() ;
; ![]()
La validité de ces équations dépend, pour la précision
voulue, essentiellement du domaine de variation de la pression.
Ainsi jusquà 2 atmosphères, et même 10 ou 20,
on utilise léquation détat des gaz parfaits.
Covolume
Léquation ![]() sécrit
sécrit ![]()
Si la pression augmente énormément à une température
donnée, le volume V tend vers la valeur B(T) = nb(T)
qui, suivant lhypothèse atomique, représente le volume occupé
par les molécules du gaz " entassées " les unes sur les autres.
Ce volume limite est appelé covolume.
Compte tenu de cette interprétation, il est logique que b
soit pratiquement indépendant de la température.
Equation détat de Van der Waals
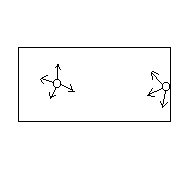 |
Dans un fluide réel, les interactions
entre molécules ne sont plus négligeables et une molécule
frappant une paroi est ralentie à linstant du choc par la présence
des autres molécules toutes situées dun même coté.
Ainsi le choc sera moins important que dans le cas du gaz parfait. Le terme p, représentant la pression dans léquation détat des gaz parfaits, doit être remplacé par p + p où p est la pression du gaz réel et p un terme devenant nul lorsque la pression tend vers 0 cest à dire lorsque le comportement du gaz réel tend vers celui du gaz parfait. |
Ainsi, une équation détat assez générale pour
un fluide réel est ![]()
Ainsi dans léquation détat de Van der Waals, et on obtient ![]() avec
avec![]() (résultats
expérimentaux).
(résultats
expérimentaux).
Cette équation de Van der Waals interprète la température
de Mariotte.
![]()
Pour ![]() , léquation
de Van der Waals sapproche au mieux de léquation détat des
gaz parfaits.
, léquation
de Van der Waals sapproche au mieux de léquation détat des
gaz parfaits.
Elle interprète aussi la température critique (chapitre
2).
On utilise dautres équations détat de type Van
der Waals, par exemple
Diétérici ![]() ; Berthelot
; Berthelot ![]() ;
;
Clausius ![]()
Les équations de Van der Waals, Diétérici, Berthelot ou Clausius peuvent, dans certaines limites, sappliquer aux liquides.
La calorimétrie a pour objet la mesure des capacités calorifiques et des chaleurs latentes de changement détat.
2.1. Mesure des capacités calorifiques
2.1.1. Méthodes de mesure pour les solides, les liquides
En réalisant une transformation isobare, on détermine expérimentalement
Cp . On calcule CV
à partir de ![]() .
.
En fait les écarts relatifs ![]() restent faibles, 5% pour laluminium, 4% pour lor, 3% pour le cuivre et largent,
... de lordre de grandeur des erreurs de mesures.
restent faibles, 5% pour laluminium, 4% pour lor, 3% pour le cuivre et largent,
... de lordre de grandeur des erreurs de mesures.
La méthode des mélanges
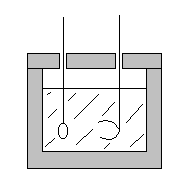 |
Un vase est rendu pratiquement adiabatique par une
enveloppe isolante (ou un système disolation complexe). Un tel système
est appelé calorimètre adiabatique. On y introduit
une masse deau de capacité calorifique massique à pression
constante Soit |
On plonge le solide de capacité calorifique massique à pression
constante ![]() dans le calorimètre le plus rapidement possible et on referme le couvercle.
dans le calorimètre le plus rapidement possible et on referme le couvercle.
On agite afin datteindre un équilibre à température finale ![]() au plus vite.
au plus vite.
Soit m la masse deau qui échangerait
dun point de vue thermique comme le vase et les accessoires (agitateur,
capteur de température).
La transformation est irréversible, isobare à la pression ![]() de lenvironnement (le plus souvent,
de lenvironnement (le plus souvent,![]() ).
).
![]()
Pour le système, corps solide + calorimètre, la quantité
de chaleur, donc la variation denthalpie, est nulle.
![]()
Lenthalpie est une fonction détat, sa variation ne dépend
que des états initial et final.
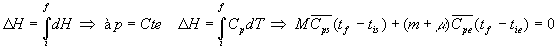
soit ![]()
Cette méthode nécessite la mesure préalable de m qui pourra être faite par la méthode électrique exposée ci-après ou en faisant une expérience préalable avec une masse m deau à température connue à la place du solide.
La méthode électrique
Particulièrement adaptée pour les liquides, elle consiste
à chauffer le liquide enfermé dans le calorimètre
adiabatique à laide dune résistance électrique.
La connaissance de lénergie électrique fournie et de lélévation
de température permet la mesure de la capacité calorifique.
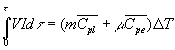
V : tension aux bornes de la résistance ; I : courant
électrique ; m : masse du liquide ; ![]() capacité calorifique massique à pression constante du liquide
; m : valeur en eau du calorimètre;
capacité calorifique massique à pression constante du liquide
; m : valeur en eau du calorimètre; ![]() :
capacité calorifique massique à pression constante de leau ;
DT : élévation de température.
:
capacité calorifique massique à pression constante de leau ;
DT : élévation de température.
Remarques :
- un calorimètre nest jamais parfaitement adiabatique (des
corrections sont nécessaires faisant appel aux transferts thermiques),
- dans les microcalorimètres, on nempêche pas les pertes
thermiques, on les favorise mais on en tient compte avec précision,
- il sagit, sur lintervalle de température, des valeurs moyennes
des capacités calorifiques doù lintérêt de
travailler pour des écarts de température les plus faibles
possibles,
- en Transferts Thermiques, dautres méthodes existent (hors
programme).
2.1.2. Méthodes de mesure pour les gaz
Ces mesures sont difficiles en raison de la faiblesse des masses volumiques
des gaz et de lobligation de les enfermer dans des calorimètres
dont la capacité calorifique est souvent plus importante que celle
du gaz.
On mesure ![]() et lon déduit
et lon déduit ![]() soit de la formule
soit de la formule ![]() ,
soit de la mesure de
,
soit de la mesure de![]() .
.
Une méthode de mesure de ![]() dun gaz est connue sous le nom de méthode du courant stationnaire.
Elle est adaptée aux liquides.
dun gaz est connue sous le nom de méthode du courant stationnaire.
Elle est adaptée aux liquides.
Une méthode de mesure de g
est connue sous le nom dexpérience de Clément et Desormes.
Ces deux méthodes sont étudiées en Travaux Pratiques.
Un autre processus de mesure de g
porte le nom de formule de Reech.
Nous avons introduit le coefficient de compressibilité isotherme![]() ,
nous introduisons celui de compressibilité adiabatique
,
nous introduisons celui de compressibilité adiabatique ![]() et remarquons que
et remarquons que ![]() en un même état.
en un même état.
![]()
![]()
Ainsi ![]()
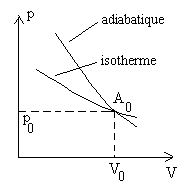 |
Traçons dans un diagramme V, p (diagramme
de Clapeyron) lisotherme passant par le point A0
de coordonnées (p0 , V0).
Les dérivées En un même point, la pente de ladiabatique, dans un diagramme V, p, est toujours plus accentuée que celle de lisotherme. |
2.1.3. Résultats
Cas des solides
Le tableau ci-dessous donne la capacité calorifique massique et molaire à pression constante pour différents corps simples à température et pression ordinaires ainsi que les masses molaires M.
|
|
|
|
|
|
|
|
|
|
|
|
| M en g |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
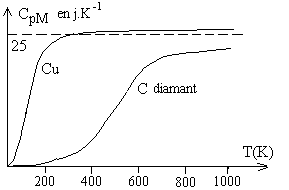 |
En fait Si la température est suffisante, |
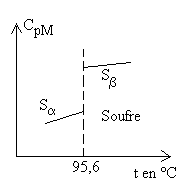 |
Certaines substances solides présentent
plusieurs structures cristallines (on dit aussi plusieurs variétés
allotropiques) et, à lintérieur de létat solide,
il y a changement de phase ou détat.
Lexpérience montre que La mesure de |
Cas des liquides
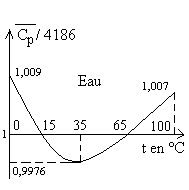 |
Il ny a pas de loi simple. Pour de nombreux liquides,
on trouve un |
Cas des gaz
Le tableau ci-après et les courbes représentées
rassemblent les principaux résultats.
|
|
|
CpM - CVM |
|
||||||||||||
|
|
|
|
|
|
|
|
|||||||||
| monoatomique He |
|
|
|
|
|
|
|
||||||||
| monoatomique Ar |
|
|
|
|
|
|
|
||||||||
| diatomique O2 |
|
|
|
|
|
|
|
||||||||
| diatomique N2 |
|
|
|
|
|
|
|
||||||||
| diatomiqueH2 |
|
|
|
|
|
|
|
||||||||
| autre gaz H2O |
|
|
|
||||||||||||
| autre gaz CO2 |
|
|
|
|
|
|
|
||||||||
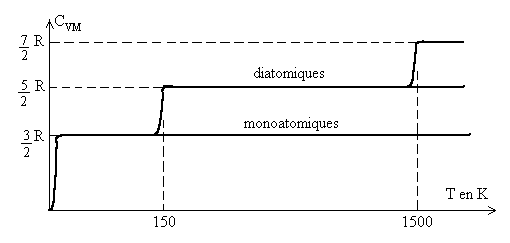
Commentaires : Les résultats sont donnés pour une
pression atmosphérique normale pour laquelle, en première
approximation, les gaz réels ont un comportement de gaz parfait.
Les courbes traduisent les évolutions de ![]() avec la température, des différences apparaissent avec latomicité.
avec la température, des différences apparaissent avec latomicité.
La valeur de ![]() est égale à R quelque soit le gaz.
est égale à R quelque soit le gaz.
A la température T = 0 K, la valeur de ![]() est nulle. Elle évolue très rapidement (entre 0 et 1K)
vers la valeur
est nulle. Elle évolue très rapidement (entre 0 et 1K)
vers la valeur ![]() quelque soit le gaz.
quelque soit le gaz.
Les gaz monoatomiques gardent cette dernière valeur quelque
soit la température.
Les autres gaz subissent deux évolutions pour ![]() ,
lune autour de 150 K, lautre autour de 1500 K.
,
lune autour de 150 K, lautre autour de 1500 K.
Pour les diatomiques, ![]() atteint la valeur
atteint la valeur ![]() ,
puis
,
puis ![]() . Pour
une atomicité supérieure, les résultats sont plus complexes.
Ils dépendent de la forme de la molécule et de son atomicité.
On atteint dabord
. Pour
une atomicité supérieure, les résultats sont plus complexes.
Ils dépendent de la forme de la molécule et de son atomicité.
On atteint dabord ![]() ,
puis une valeur plus élevée.
,
puis une valeur plus élevée.
2.2. Chaleurs latentes de changement de phase
2.2.1. Définition
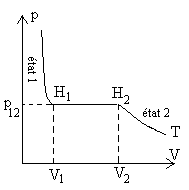 |
A pression constante, la variation denthalpie
est égale à la quantité de chaleur échangée
dans la transformation.
On appelle chaleur latente (massique ou molaire) de changement de phase dun corps pur à la température T la variation denthalpie (de lunité de masse ou dune mole) de ce corps passant dun état (solide, liquide ou gazeux) à un autre état. |
La notation habituelle pour les chaleurs latentes est L. Ainsi ![]() , la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
, la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
2.2.2. Mesure des chaleurs latentes
Chaleur latente de fusion
On utilise un calorimètre adiabatique dans lequel est maintenu à
une température constante ![]() légèrement supérieure à la température de
fusion
légèrement supérieure à la température de
fusion ![]() du
corps à étudier. On y introduit une masse m de ce corps
à une température
du
corps à étudier. On y introduit une masse m de ce corps
à une température ![]() légèrement inférieure à
légèrement inférieure à ![]() (le corps est donc en phase solide) ; on maintient la température du
calorimètre constante à laide dune résistance immergée
dans le calorimètre et parcourue par un courant électrique réglable.
(le corps est donc en phase solide) ; on maintient la température du
calorimètre constante à laide dune résistance immergée
dans le calorimètre et parcourue par un courant électrique réglable.
![]() , si Q
est la quantité de chaleur fournie par effet Joule,
, si Q
est la quantité de chaleur fournie par effet Joule, ![]() les capacités calorifiques massiques du corps à létat
solide et liquide et
les capacités calorifiques massiques du corps à létat
solide et liquide et ![]() la chaleur latente massique de fusion du corps.
la chaleur latente massique de fusion du corps.
Chaleur latente de vaporisation
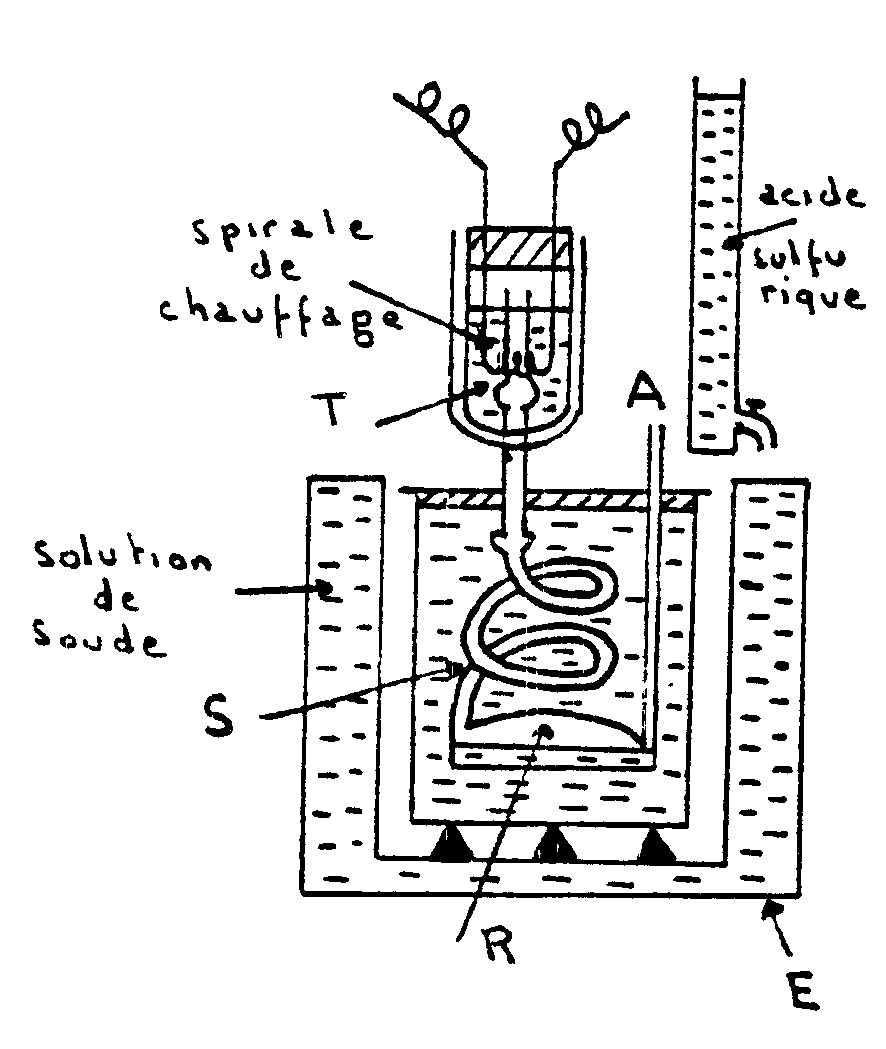
Appareil de Richards |
Un vase Dewar est traversé par un tube
T ouvert à ses deux extrémités. Ce tube se raccorde
à un serpentin S plongé dans un calorimètre adiabatique.
Le serpentin aboutit à un réservoir de condensation R et
communique avec latmosphère par le tube A . Lébullition
du liquide placé dans le vase Dewar a lieu ainsi sous la pression
atmosphérique.
Des gouttes liquides peuvent provenir dune légère condensation qui se produit dans la vapeur en montant dans le vase Dewar. Elles sont vaporisées à nouveau lorsquelles traversent le fond dans le tube T et la vapeur arrivant dans S est sèche. Légalité de température entre le calorimètre et lenceinte E est obtenue en faisant tomber de lacide dans la solution de soude contenue dans E. |
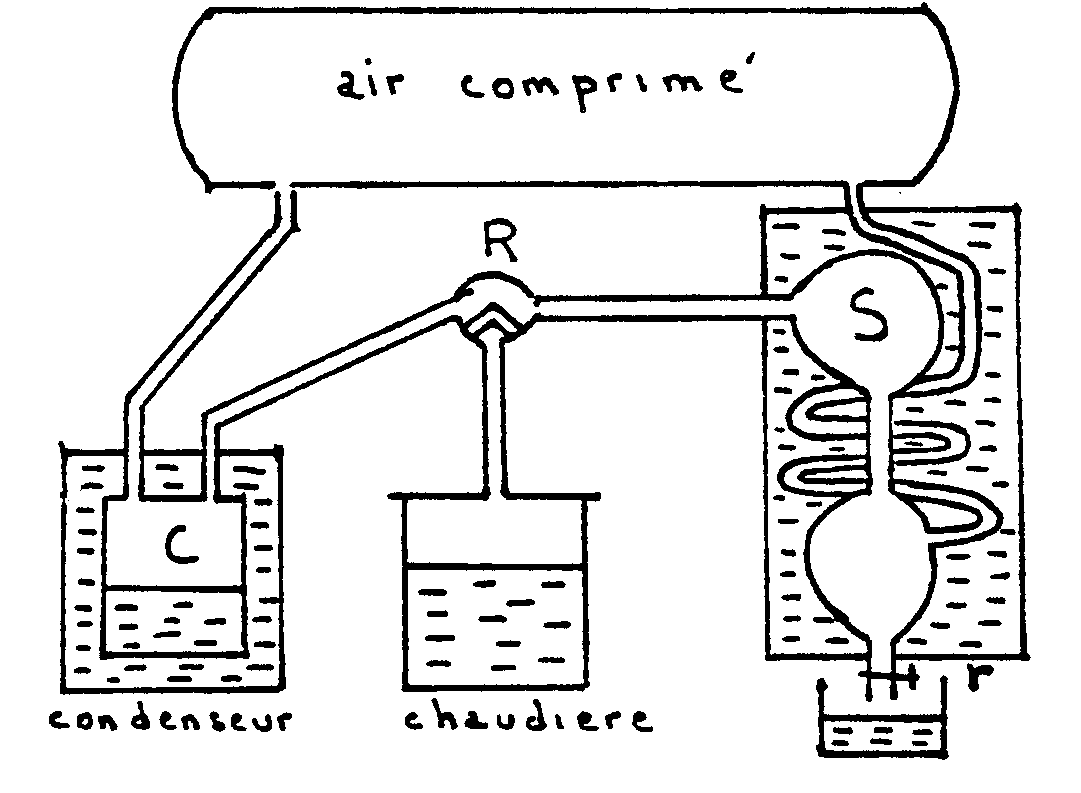 |
Lappareil est entièrement clos et la
pression intérieure est obtenue grâce à une atmosphère
artificielle (réservoir de 600 litres pouvant supporter des pressions
inférieures à 20 atmosphères). Un robinet R permet
de mettre la chaudière en communication soit avec un condenseur
C, soit avec un serpentin de condensation S.
Au début, la chaudière communique avec le condenseur et on chauffe le liquide (eau) jusquà ébullition ; les vapeurs viennent se condenser dans C. |
Quand la distillation est devenue régulière, on met la
chaudière en communication avec le serpentin. La vapeur sèche
qui arrive sy condense et quand lexpérience a duré assez
longtemps, on remet la chaudière en communication avec le condenseur,
puis on recueille avec le robinet r leau condensée en S.
Désignons par m la masse du liquide condensé, ![]() sa capacité calorifique massique,
sa capacité calorifique massique, ![]() sa chaleur latente de vaporisation sous la pression considérée
à la température
sa chaleur latente de vaporisation sous la pression considérée
à la température ![]() et
et ![]() la valeur
en eau du calorimètre dont la température initiale est
la valeur
en eau du calorimètre dont la température initiale est ![]() et la température finale
et la température finale ![]() .
.
Le bilan énergétique sécrit : ![]()
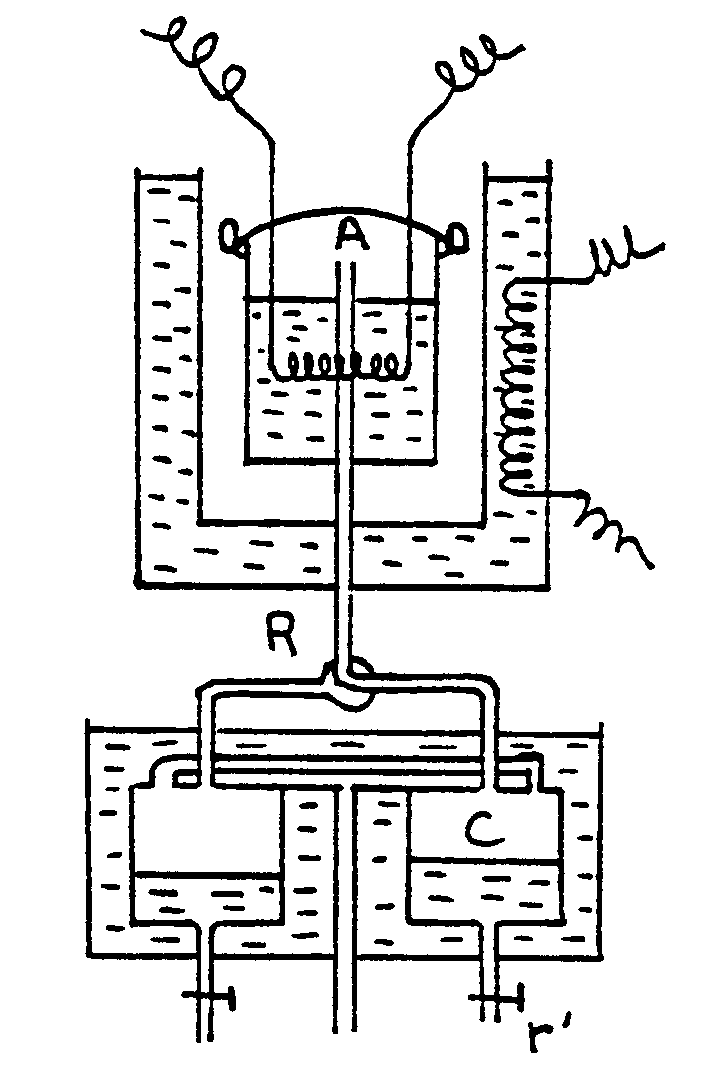 |
On produit la vaporisation par le passage dun
courant dintensité I dans une résistance R
immergée dans le liquide.
Si, pendant le temps La vaporisation est produite dans une enceinte close A, très robuste. La vapeur se dégage par un ajustage muni dun robinet R. Elle se condense dabord dans un condenseur auxiliaire jusquà ce que lon obtienne un régime permanent. La vapeur est ensuite condensées dans C, recueillie par le robinet r et pesée. Soit m la masse du liquide vaporisé, m celle de la vapeur qui est sortie de la chaudière (que lon a recueillie par condensation), |
Laugmentation de volume ![]() produite par la vaporisation est égale au volume de vapeur
produite par la vaporisation est égale au volume de vapeur ![]() sorti
de la chaudière ð
sorti
de la chaudière ð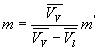 .
.
m diffère dautant plus de m que lécart ![]() est plus faible cest à dire que lon sapproche de la température
critique.
est plus faible cest à dire que lon sapproche de la température
critique.
Le tableau ci-après donne les valeurs pour leau.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.2.3. Résultats
Chaleur latente de fusion
Sous la pression atmosphérique, pour différents corps :
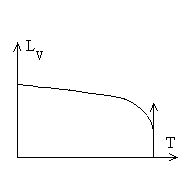 |
Les chaleurs latentes de vaporisation diminuent
avec la température et atteignent une valeur nulle au point critique
puisque phases liquide et gazeuse sont identiques.
On utilise souvent des lois empiriques du type Ainsi pour leau entre 100 et 200 °C, |
Nous donnons ci-après quelques chaleurs latentes massiques de vaporisation exprimées en kj/kg sous une pression dun atmosphère.
Ammoniac 1425 ; Benzène 393 ; Dioxyde de Carbone 594 ; Dioxyde de Soufre 397 ; Eau 2253 ; Ethanol 903
Limportances des valeurs des chaleurs latentes et, plus particulièrement, de celles de vaporisation justifie lutilisation des changements de phase dans beaucoup de machines thermiques.
Les chaleurs latentes de fusion, de sublimation et de vaporisation sont positives, celles de solidification, de condensation à létat solide et de condensation à létat liquide sont respectivement égales et opposées.