Chapitre 2 : Structure de la matière
Plan
1. Introduction : gaz, liquide, solide
2. Les particules de base de la matière
2.1. Lhypothèse atomique (ou moléculaire)
2.2. La particule de base
3. Les deux états de la matière : létat
désordonné et létat ordonné
3.1. Létat ordonné
3.1.1. Agitation au niveau microscopique
3.1.2. Interprétation microscopique de la
pression
3.1.3. Facteur de Boltzmann. Raréfaction de
lair
3.1.4. Température cinétique. Equipartition
de lénergie
3.1.5. Du gaz comprimé au liquide
3.1.6. Agitation thermique dans les liquides. Viscosité
des liquides
3.2. Létat ordonné : le solide cristallisé
4. Changements de phases des corps purs
4.1. Introduction
4.2. Observation courante
4.3. Equilibre liquide-vapeur
4.4. Equilibre liquide-solide, équilibre solide-gaz
4.5. Remarque
Annexe : la loi de distribution des vitesses de Maxwell
Physiciens et chimistes étudient les propriétés
de la matière. Leurs domaines respectifs ne sont pas réservés,
ils appartiennent à un même domaine scientifique appelé
Sciences
Physiques. Les collègues de lenseignement secondaire sont professeurs
de Physique et Chimie. Parce que le domaine de connaissances en Sciences
Physiques croît de manière exponentielle, lenseignement supérieur
a été amené à distinguer des professeurs de
Physique et des professeurs de Chimie sans quil y ait une frontière
réelle, tangible entre eux.
Traditionnellement, les premières notions datomistique,
de liaisons et dédifices entre les atomes font partie des leçons
de Chimie de lenseignement secondaire.
Nous ne croyons pas que lon puisse faire des études de Physique
en ignorant ces pré-requis.
1. Introduction : gaz, liquide, solide
Communément, on classe les milieux matériels en solides
et fluides.
Ces deux notions, comme beaucoup de notions premières, ne sont
pas simples à définir, la difficulté augmente si l'on
souhaite, comme tout un chacun, distinguer les liquides et les gaz à
l'intérieur de la famille fluide.
Remarquons cependant que tout au long du premier chapitre nous avons utilisé notre sens commun pour nous représenter la matière et avons pu, sans trop de difficulté de compréhension entre nous, donner un contenu scientifique à des notions aussi tangibles que pression ou température.
Il nest jamais inutile douvrir un dictionnaire et de chercher le sens
qui peut être donné aux mots.
Ouvrons (sans en faire publicité) le "Petit Robert" et regardons
les définitions à usage courant.
Fluide : "Qui n'est ni solide, ni épais, coule aisément".
Mais l'huile, par exemple, c'est épais ou non ? Allons voir à
solide.
Solide : "Qui a de la consistance, qui n'est pas liquide, tout
en pouvant être plus ou moins mou". Qui n'est pas liquide ? Allons
voir à liquide.
Liquide : "Tout corps qui coule ou tend à couler". Peut-on
dire que le sable sec qui coule entre les doigts est un liquide ?
Les définitions à usage courant semblent bien floues, aussi dans le même dictionnaire nous considérons les définitions scientifiques.
Fluide : "Tout corps qui se laisse déformer sous l'action
de forces minimes; tout corps qui épouse la forme de son contenant"
Solide : "Se dit d'un corps dans lequel les molécules
sont très rapprochées les unes des autres et vibrent avec
une très faible amplitude autour de leur position d'équilibre;
qui a de la cohésion, garde une forme relativement constante lorsqu'il
n'est pas soumis à des forces extérieures"
Liquide : "Tout corps à l'état fluide, pratiquement
incompressible, formé de corpuscules (ions ou molécules)
soumis à de faibles attractions"
Gaz : "Tout corps qui se présente à l'état
de fluide expansible et compressible (état gazeux) dans les conditions
de température et de pression normales"
Cette lecture permet de dégager quelques pistes :
2. Les particules de base de la matière
2.1. Lhypothèse atomique (ou moléculaire)
Lhypothèse est banale (universellement admise). Elle était
déjà émise par les philosophes de la Grèce
antique. Par contre les premières preuves expérimentales
- Dalton, Proust, Lavoisier au 19ème
siècle - sont relativement récentes.
Si, dans un cataclysme, toute notre connaissance scientifique devait
être détruite et quune seule phrase passe aux générations
futures, quelle affirmation contiendrait le maximum dinformation dans
le minimum de mots ? Nest ce pas lhypothèse atomique à
savoir " toutes les choses sont faites datomes (de molécules),
petites particules en agitation incessante, sattirant mutuellement à
petite distance les unes des autres et se repoussant si on cherche à
trop les rapprocher ". Dans cette seule phrase vous verrez quil y a une
énorme quantité dinformations sur le monde, si on lui applique
un petit peu dimagination et de réflexion. The Feynman Lectures
on Physics par Richard P. Feynman, Robert B. Leighton et Mattews Sands.
Sans doute, cette phrase de Richard P. Feynman, prix Nobel de Physique en 1965, est suffisante pour aborder un premier cours de Thermodynamique à condition de la compléter par :
La compressibilité des gaz, la presque incompressibilité
des liquides et des solides permet de dire que les atomes dun liquide
ou dun solide sont " entassés " les uns sur les autres alors que
ceux dun gaz sont éloignés les uns des autres. Ceci est
confirmé par la masse volumique qui est faible (peu datomes par
unité de volume) pour les gaz et importante pour les liquides et
les solides, par le fait que nous nous sentons " léger " (forte
poussée dArchimède) dans leau et " lourd " dans lair,
que nous avons du mal à marcher dans leau,
Létat gazeux est un état dilué
alors que liquides et solides font partie des états condensés.
2.2.1. L'atome
Toute matière est constituée d'atomes.
L'atome est constitué d'un noyau comportant Z protons et d'un
nombre au moins égal de neutrons (à l'exception de l'atome
d'hydrogène qui ne comporte pas de neutron), de Z électrons
gravitant autour du noyau.
En Chimie, on apprend quil existe une bonne centaine datomes " libres
". Le tableau de Mendeleïev les regroupe de manière ordonnée
suivant leurs caractéristiques. Les théories quantiques actuellement
admises ont considérablement perfectionné ce modèle
atomique élémentaire rappelé ici.
La masse du proton est égale à ![]() et sa charge
et sa charge ![]() .
.
La masse du neutron est égale à ![]() ,
il est électriquement neutre.
,
il est électriquement neutre.
La masse de l'électron est égale à ![]() et sa charge est égale et opposée à celle du proton.
et sa charge est égale et opposée à celle du proton.
Chacun des Z électrons est dans un " état " caractérisé,
entre autres, par l'énergie qu'il faut dépenser pour le libérer
sans vitesse appréciable de l'atome. Les valeurs de ces énergies
permettent de classer les Z électrons en " couches " de même
énergie où le nombre possible d'électrons est
limité. Les niveaux dénergie entre couches sont nettement
séparées au moins pour les plus fortes valeurs.
On distingue ces couches en introduisant un nombre entier positif n appelé
nombre quantique principal. Ainsi, la couche la plus profonde (première
à partir du noyau) appelée K correspond à ![]() ,
la seconde L à
,
la seconde L à ![]() et ainsi de suite.
et ainsi de suite.
Pour latome dhydrogène (et pour les ions hydrogénoïdes
qui, comme latome dhydrogène ne possèdent quun électron),
lénergie de lélectron suivant la couche où il se trouve
est égale à : ![]() .
.
Pour des atomes ou des ions polyélectroniques, les règles
de Slater pour calculer lénergie dun électron dune couche
constituent une bonne approximation empirique.
Pour ![]() , la
formule ci-dessus reste valide ; pour l'Uranium (
, la
formule ci-dessus reste valide ; pour l'Uranium (![]() ),
les mesures donnent 115600 eV.
),
les mesures donnent 115600 eV.
Les électrons dune couche " tournent " sur des orbites définies
par les nombres quantiques secondaire et magnétique.
2.2.2. Les particules de base
Etablir un modèle de structure de la matière consiste
à décrire l'arrangement des atomes, en représentant
leurs positions respectives et la répartition des différentes
espèces. Ce qu'on pourrait imaginer de plus simple serait que les
atomes se comportent comme des constituants, doués de propriétés
spécifiques, parfaitement définis et immuables. Ce nest
pas aussi simple puisque les atomes apparaissent dans la matière
avec des petites différences par rapport au schéma de l'atome
libre qui fait l'objet du tableau de Mendeleïev. Ces petites différences
sont essentielles pour rendre compte des propriétés de la
matière.
En fait, dans une transformation physique ou une réaction chimique,
seul l'état des quelques électrons externes peut être
modifié. La ménagère sur sa cuisinière, le
chimiste dans son laboratoire, le métallurgiste avec son haut-fourneau
ou son four à arc ne peut qu'égratigner les atomes. Avec
une excellente approximation, le noyau et tous les électrons
sauf les quelques uns des couches externes sont immuables.
Toute matière est faite d'un ensemble " d'entités ",
comme une construction est faite de briques, différentes suivant
la matière. Nous désignons ces entités par le terme
"
particule " ou mieux " particule de base ". Dans quelques cas
rares ce sont des atomes tels qu'ils sont à l'état libre,
plus souvent des atomes modifiés par l'altération des couches
électroniques externes ou des groupes d'atomes définis appelés
" molécules ".
La " particule de base " est représentée par un objet
de forme et de dimensions déterminées et le modèle
de la matière est l'assemblage de ces objets. Ainsi un atome est
représenté par une sphère de diamètre donné
et un ensemble d'atomes par de telles sphères pressées les
unes contre les autres. A priori cette représentation n'est pas
évidente mais elle apporte une image simplificatrice primordiale
pour notre compréhension de la structure de la matière.
La molécule mono et polyatomique
Les gaz rares sont caractérisés par une couche électronique
externe complète (qui ne peut accepter un électron de plus).
Cette propriété les fait bénéficier d'une stabilité
toute particulière qui les rend pratiquement inerte et la particule
de base est faite d'un atome non altéré. On parle de molécules
monoatomiques.
Les atomes, autres que les gaz rares, se lient entre eux suivant des
schémas bien définis formant un ensemble stable appelé
molécule
polyatomique. La liaison interatomique provoque l'altération
de l'état des électrons des couches externes. Décrire
les modes de liaisons et les mécanismes des " réactions "
relève du domaine de la Chimie, le minimum à connaître
étant les règles de " remplissage ".
Dans la matière à l'état dilué (gaz),
la molécule (à lexception des plasma qui sont constitués
dions et délectrons) est la particule de base.
Dans celle à l'état condensé (liquide et solide),
Ce sont bien des atomes mais ils n'ont pas exactement la même
constitution que l'atome de même espèce à l'état
isolé. En effet, quand la matière est condensée, cela
signifie que chaque atome se trouve au contact d'autres, donc qu'il est
soumis à des interactions qui altèrent l'état des
électrons de la couche superficielle.
C'est ce qui se passe pour les atomes formant les molécules,
la différence essentielle est que, dans la molécule, les
atomes constituants forment un système fini, bien défini,
auquel on ne peut adjoindre ou retirer un atome sans modifier profondément
la molécule. Au contraire, dans un solide dont les éléments
constitutifs sont des atomes, ceux-ci forment des chaînes en interaction
non finies.
Nous illustrons notre propos avec l'atome de Carbone qui possède
6 électrons, 2 sur sa première couche (K) qui est complète
et quatre sur sa deuxième couche (L) externe qui peut être
complétée à 8.
Avec 4 atomes d'Hydrogène, le Carbone forme la molécule ![]() ,
ensemble fini, où le Carbone partage les 4 électrons des atomes
d'Hydrogène pour compléter sa couche externe, où chaque
atome d'Hydrogène partage 1 électron de l'atome de Carbone pour
compléter sa seule couche à 2 électrons.
,
ensemble fini, où le Carbone partage les 4 électrons des atomes
d'Hydrogène pour compléter sa couche externe, où chaque
atome d'Hydrogène partage 1 électron de l'atome de Carbone pour
compléter sa seule couche à 2 électrons.
Dans le solide diamant, un atome de Carbone partage 1 électron
avec chacun des 4 atomes de Carbone qui l'entourent pour compléter
sa couche externe, ces derniers en faisant de même si bien que les
atomes du diamant forment des chaînes bien déterminées
qui se développent dans toutes les directions. Il n'y a pas de molécule
de diamant. En ce qui concerne les atomes de Carbone à la surface
du diamant, ils complètent leur couche externe avec des impuretés
de l'atmosphère environnante pour assurer la stabilité de
l'édifice diamant.
Les ions, éléments de la matière condensée
Un atome, dont le nombre atomique est supérieur de quelques unités
(1, 2, 3, voire 4) à celui d'un gaz rare, retrouve la configuration
électronique de celui-ci s'il perd ses électrons extérieurs.
Cette opération, qui nécessite peu d'énergie,
explique lexistence des solides et liquides métalliques . L'atome
n'est plus électriquement neutre : il devient un ion chargé
positivement -cation- de 1, 2, ... charges élémentaires,
environné dun nombre égal délectrons " libres ".
Lexistence de charges positives et négatives, qui sattirent, donnent
une cohésion à lassemblage.
Inversement, si l'atome à 1, 2, ... électrons de moins
qu'un gaz rare, des électrons libres (à énergie nulle)
peuvent venir occuper des places vacantes de la couche externe de niveau
d'énergie inférieure. L'atome devient un ion chargé
négativement -anion-.
La formation dions de nature opposée est spontanée si
le bilan énergétique est exothermique.
Exemple : 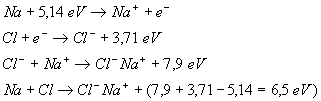
Il convient de bien comprendre.
A létat gazeux il existe une molécule (dans lexemple![]() ).
Pour ce qui est de létat solide ou liquide, il sagit dun ensemble
d'ions positifs et négatifs (dans lexemple
).
Pour ce qui est de létat solide ou liquide, il sagit dun ensemble
d'ions positifs et négatifs (dans lexemple ![]() )
qui, en sattirant, assurent la cohésion de lédifice. Dans cet
ensemble, il n'y a pas de couples isolés (
)
qui, en sattirant, assurent la cohésion de lédifice. Dans cet
ensemble, il n'y a pas de couples isolés (![]() ),
donc pas de molécule : la particule de base du modèle est bien
l'ion, c'est à dire un atome modifié dans sa couche extérieure
par la perte ou le gain d'un petit nombre d'électrons.
),
donc pas de molécule : la particule de base du modèle est bien
l'ion, c'est à dire un atome modifié dans sa couche extérieure
par la perte ou le gain d'un petit nombre d'électrons.
Nos exemples définissent l'ion à partir d'un atome. Il
existe des ions plus complexes constitués de plusieurs atomes liés
entre eux comme dans une molécule, la charge des électrons
et des noyaux ne se compensant pas totalement.
![]()
La dimension des ions, atomes et molécules
La masse des particules ne pose pas de problème particulier.
Dans l'atome, toute la masse est pratiquement concentrée dans le
noyau à environ 1/4000 près ou moins. La modification de
l'état des électrons, et de légers changements dans
leur nombre, ne font pas varier la masse qui est simplement celle du ou
des noyaux constituant la particule.
Les dimensions géométriques sont plus délicates à
définir. Dans l'image qu'en donne la mécanique quantique, l'atome
libre est constitué par un nuage d'électricité négative,
de charge totale ![]() ,
autour du noyau ponctuel de charge
,
autour du noyau ponctuel de charge ![]() .
La densité locale de charge est calculable par les équations de
la mécanique quantique.
.
La densité locale de charge est calculable par les équations de
la mécanique quantique.
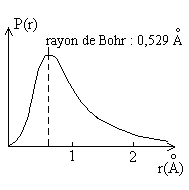 |
Pour l'atome d'Hydrogène qui peut être
calculé rigoureusement, la figure ci-contre donne la probabilité
de présence de l'électron à une distance r.
Elle est maximale pour la valeur du rayon de Bohr, égale à 50% pour r = 0,707 Å, à 90% pour r = 1,408 Å. |
La situation est très différente dans les ensembles denses d'atomes qu'on observe dans la matière condensée, solide ou liquide.
Ce que nous venons d'expliquer est très simplifié.
En particulier, rien ne nous permet de comprendre ce qui empêche
deux atomes en interaction de se pénétrer jusqu'aux noyaux
puisqu'ils sont essentiellement constitués de vide. Il faut, pour
cela, abandonner les concepts classiques pour la mécanique quantique
et introduire les interactions nucléaires.
3. Les deux états de la matière : l'état désordonné et l'état ordonné
Le sens commun nous fait distinguer trois états de la matière
: solide, liquide et gazeux ... sans que nous soyons capables d'en faire
les distinguo de manière rigoureuse.
Le modèle atomique de la matière conduit à une
séparation des états physiques en deux classes dont le mérite
est une coupure plus franche : le première classe est dite à
structure
désordonnée, la deuxième à
structure
ordonnée.
3.1. L'état désordonné : du gaz au liquide
3.1.1. Agitation au niveau microscopique
Les modèles ne sont jamais statiques (ce qui n'est pas en contradiction
avec " statique " ou " globalement au repos " au niveau macroscopique).
C'est particulièrement évident pour un gaz où
la matière est diluée cest à dire où les particules
sont " libres " de leur déplacement. En effet, si elles n'étaient
pas en mouvement, du fait du champ de pesanteur, elles ne pourraient être
qu'entassées à partir du fond du récipient qui les
contient ou à partir du sol ce qui ne peut manifestement être
le cas pour une substance diluée.
Ainsi dans les gaz, sintroduit la notion de mouvement sans cesse des
particules, dans toutes les directions au gré des chocs, que l'on
appelle agitation. Les particules seront attirées vers les
altitudes les plus faibles mais sans pouvoir s'y entasser.
Dans un gaz, les interactions entre particules sont faibles (matière
diluée, particules en moyenne éloignées les unes des
autres), les mouvements de celles-ci sont assez libres, il règne
un désordre, que l'on qualifie de parfait si on néglige
toute interaction entre particules.
La théorie cinétique des gaz lie agitation
et température, c'est pourquoi on parle d'agitation thermique.
3.1.2. Interprétation microscopique de la pression
Une conséquence de l'agitation est une visualisation locale (au
niveau microscopique) de la pression.
La pression est la force moyenne par unité de surface due aux
particules venant frapper une paroi.
La pression augmente :
- avec le nombre de chocs contre la paroi donc, dans une enceinte de
volume V, avec le nombre N de particules (soit, en fait, avec N/V),
- avec l'agitation des particules puisque la quantité de mouvement
transférée de la particule à la paroi sera d'autant
plus importante.
Notons l'accord entre le caractère non directionnel de la pression
et le fait que le mouvement des particules ait lieu dans toutes les directions.
De même pour que la pression tende vers une valeur nulle (comportement
limite de tous les gaz réels appelé gaz parfait), il convient
que le nombre de chocs soient faible, cest à dire que la densité
des particules soit faible, cest à dire quen moyenne elles soient
éloignées les unes des autres, donc que les interactions
entre les particules soient négligeables.
3.1.3. Facteur de Boltzmann. Raréfaction de l'air
Nous traitons là un exemple de statique des fluides compressibles,
lair atmosphérique obéissant à léquation
détat des gaz parfaits.
Nous cherchons à savoir comment varie la pression p avec
laltitude z. Nous formulons lhypothèse justifiée
que lair est un mélange idéal de gaz parfaits et celle fausse
dans la troposphère (altitude comprise entre 0 et 20
km) que la température est la même quelque soit laltitude.
Soient ![]() la
température et
la
température et ![]() la masse molaire de lair.
la masse molaire de lair.
Nous utilisons le principe de lhydrostatique dp = -mg
dz et léquation détat des gaz parfaits.
![]()
![]()
A température uniforme, la pression est proportionnelle à
la masse volumique ainsi quà la densité volumique molaire.
On obtient :
![]()
![]()
où m = Ma /N représente la masse " moyenne " dune molécule dair.
![]() est lénergie
potentielle dans le champ de pesanteur terrestre de la " molécule dair
" à laltitude z.
est lénergie
potentielle dans le champ de pesanteur terrestre de la " molécule dair
" à laltitude z.
Cette application nous explique le phénomène de raréfaction
de lair avec laltitude, à savoir la densité moléculaire
diminue avec laltitude suivant une fonction dépendant de lénergie
de la molécule à cette altitude et de la température.
La fonction ![]() appelée facteur de Boltzmann est dune importance considérable
en Physique statistique.
appelée facteur de Boltzmann est dune importance considérable
en Physique statistique.
Dans l'exemple considéré, elle sinterprète de la manière
suivante : le rapport ![]() est la probabilité de présence d'une molécule à
l'altitude z. Si, pour une altitude, ce rapport est égal à 1/2,
ceci veut dire qu'il y a deux fois moins de molécules à cette
altitude qu'à l'altitude de référence ou, exprimé
différemment, que la probabilité de présence d'une molécule
à l'altitude z est 50% de celle de la trouver à l'altitude de
référence.
est la probabilité de présence d'une molécule à
l'altitude z. Si, pour une altitude, ce rapport est égal à 1/2,
ceci veut dire qu'il y a deux fois moins de molécules à cette
altitude qu'à l'altitude de référence ou, exprimé
différemment, que la probabilité de présence d'une molécule
à l'altitude z est 50% de celle de la trouver à l'altitude de
référence.
3.1.4. Température cinétique. Equipartition de l'énergie. Energie dun gaz parfait
La distribution des vitesses des centres de masse des molécules
dun gaz parfait en équilibre à température T
vérifie
le facteur de Boltzmann.
Ce résultat est connu sous le nom de loi distribution des
vitesses de Maxwell-Boltzmann et entre dans le cadre de la théorie
cinétique du gaz parfait.
Résultat des travaux de Maxwell et de Boltzmann à la
fin du 19ème siècle, elle a été
validée expérimentale par les expériences sur les
" jets monoatomiques ". Il convient de citer la première en 1947
de Estermann, Simpson et Stern et celle très convaincante réalisée
en 1959 par Marcus et McFee.
Le lecteur trouvera en annexe à la fin de ce chapitre un complément sur la théorie cinétique du gaz parfait.
Nous retiendrons deux résultats fondamentaux :
Remarque : en remplaçant dans léquation détat
du gaz parfait, on obtient ![]() relation tout à fait cohérente avec linterprétation microscopique
de la pression.
relation tout à fait cohérente avec linterprétation microscopique
de la pression.
Energie interne dun gaz parfait
Lénergie interne dun gaz parfait est constituée des énergies
cinétiques des molécules dans leur mouvement et de la somme (que
nous notons ![]() des énergies internes à chaque molécule. Les énergies
dinteraction entre molécules sont négligeables (état raréfié).
des énergies internes à chaque molécule. Les énergies
dinteraction entre molécules sont négligeables (état raréfié).
Pour un gaz monoatomique de N molécules où lénergie
cinétique des molécules est lénergiede translation
du centre de masse, on obtient : ![]() qui correspond à la décomposition dun mouvement de translation
quelconque en trois mouvements de translation indépendants (ð
3 degrés de liberté).
qui correspond à la décomposition dun mouvement de translation
quelconque en trois mouvements de translation indépendants (ð
3 degrés de liberté).
En Calorimétrie, on accède par la mesure à la quantité![]() appelé capacité calorifique à volume constant.
appelé capacité calorifique à volume constant.
Le graphique ci-après montre des résultats, pour une
mole de gaz monoatomique raréfié, tout à fait conformes
à la théorie (sauf aux très basses températures).
Pour les gaz polyatomiques raréfiés, les résultats expérimentaux
sont plus complexes : pour les diatomiques, la capacité calorifique molaire
à volume constant ![]() est nulle à 0 K, elle atteint la valeur
est nulle à 0 K, elle atteint la valeur ![]() à partir de 2-3 K, puis
à partir de 2-3 K, puis ![]() à partir de 150 K,
à partir de 150 K,![]() à très hautes températures.
à très hautes températures.
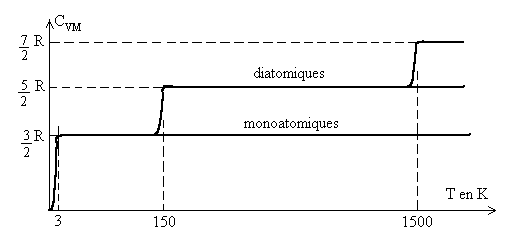
Pour expliquer ces augmentations des valeurs de la capacité calorifique,
il faut ajouter des sources dénergie : énergie de rotation
des molécules correspondant à deux degrés de liberté
(deux rotations indépendantes) et lénergie de vibration
des atomes constituant les molécules (deux degrés de liberté
correspondant à de lénergie cinétique et à
de lénergie potentielle). On trouve donc une énergie supérieure
à celle trouvée pour un gaz monoatomique.
Pour des molécules datomicité supérieure, les
résultats expérimentaux se compliquent mais linterprétation
en termes dénergie de translation, de rotation et de vibration
demeure.
Remarque : on ne peut comprendre les évolutions de ![]() avec la température sans notion de mécanique quantique et de quantum
dénergie.
avec la température sans notion de mécanique quantique et de quantum
dénergie.
3.1.5. Du gaz comprimé au liquide
Que se passe-t-il quand, sous l'action d'une pression croissante, on
atteint une densité bien plus élevée que celle des
gaz ?
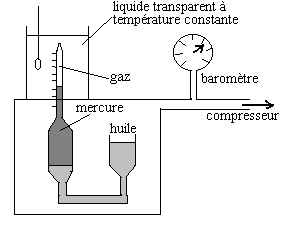 |
Du 17ème siècle jusquà
nos jours, de très nombreuses expériences de compressibilité
isotherme des gaz ont été réalisées sur tous
types de gaz, des très basses températures aux très
hautes, jusquà des pressions de 1000 atm.
Ci-contre un schéma dappareillage de compressibilité isotherme. |
Nous prenons, à titre d'exemple, la matière ![]() contenue
dans un récipient, nous utiliserons le rapport du volume moyen
contenue
dans un récipient, nous utiliserons le rapport du volume moyen ![]() occupé par la molécule au volume propre
occupé par la molécule au volume propre ![]() de la molécule
de la molécule ![]() que l'on peut évaluer à 16,8 Å3
.
que l'on peut évaluer à 16,8 Å3
.
Nous partons de l'état gazeux (A) où ![]() .
C'est typiquement un " gaz " de molécules d'eau.
.
C'est typiquement un " gaz " de molécules d'eau.
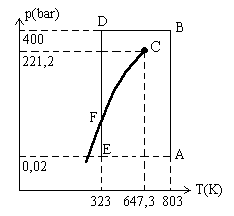 |
Nous comprimons le gaz jusqu'en (B) où Faisons une autre expérience, nous comprimons à partir de (E) où |
L'une est le gaz qui subsiste, l'autre est une phase dense (![]() ),
le liquide qui se rassemble à la partie inférieure du récipient
et qui est limitée par une surface libre horizontale au-dessous de la
phase gazeuse. Tant que coexistent les deux phases, la pression ne change pas,
il y a de plus en plus de liquide lorsque le volume diminue.
),
le liquide qui se rassemble à la partie inférieure du récipient
et qui est limitée par une surface libre horizontale au-dessous de la
phase gazeuse. Tant que coexistent les deux phases, la pression ne change pas,
il y a de plus en plus de liquide lorsque le volume diminue.
Quand toute la matière est à l'état liquide, il faut des
augmentations de pression importantes pour obtenir des diminutions de volume
faible. Ainsi en (D), ![]() .
.
Revenons à l'état (B) où le gaz est comprimé
et faisons la transformation de (B) à (D). Nous n'observons aucune
discontinuité des propriétés physiques de la matière.
Nous pensons qu'en (D) nous avons un gaz comprimé, alors que l'expérience
précédente nous avait indiqué que c'était un
liquide.
Nous concluons que liquide et gaz sont un même état
désordonné qui se distinguent par la densité.
Le liquide est représentable par un ensemble de molécules
en désordre mais serrées les unes contre les autres, comme
des billes au fond d'un sac. Il existe entre l'état gazeux et l'état
liquide une suite continue d'états intermédiaires avec un
nombre progressivement croissant de molécules par unité de
volume.
Dans la transformation ABD, les molécules de gaz se sont rapprochées
progressivement les unes des autres, la matière restant, à
chaque stade de la transformation, homogène.
Dans la transformation ED et jusqu'à atteindre l'état
(F), les molécules se rapprochent les unes des autres tout en restant
éloignées, la matière étant homogène.
A partir de ce stade, certaines molécules s'agglutinent (condensation)
formant des îlots de liquide, le reste des molécules non rassemblées
gardent la même densité et produisent la même pression.
Ces îlots fusionnent et grossissent en gouttelettes (dans le champ
de pesanteur, celles-ci tombent), le volume peut diminuer, le gaz conservant
la même pression. Dans la transformation DE, à partir de l'état
(F) les gouttelettes de liquide se désagrègent, en nombre
de plus en plus grand, formant le gaz : ce phénomène est
appelé vaporisation.
Ces explications ne nous permettent pas de comprendre l'existence d'une température
appelée critique (égale à 647,3 K dans l'exemple de l'entité
chimique ![]() ).
Il faudrait faire appel au mécanisme d'interaction entre les molécules.
).
Il faudrait faire appel au mécanisme d'interaction entre les molécules.
Par contre, ce modèle montre pourquoi un gaz est facilement
compressible alors qu'un liquide l'est très peu.
L'état gazeux à une température inférieure
à la température critique porte le nom spécifique
de vapeur. Lorsqu'il y a coexistence de vapeur et de liquide, la
pression dite pression saturante ne dépend que de la température.
On parle alors de vapeur saturante et de liquide saturant.
On remarquera que le liquide seul a une pression supérieure à
la pression saturante, que la vapeur seule (appelée vapeur sèche)
a une pression inférieure. La vapeur sèche a toutes les caractéristiques
d'un gaz. Il s'agit d'une simple distinction de langage relative au niveau
de température. Vapeur sèche et gaz ont un comportement différent
seulement au cours d'une compression isotherme.
Dans le paragraphe 53 " Equilibre liquide-vapeur ", ces résultats expérimentaux seront présentés dans un diagramme V, p.
3.1.6. Agitation thermique dans les liquides. Viscosité des liquides. Solide amorphe
Pour le liquide, comme pour tout autre état de la matière,
un modèle statique au niveau microscopique n'est pas correct car
les molécules de liquide sont soumises à l'inévitable
agitation thermique, par contre les mouvements ne sont pas libres comme
dans un gaz.
Un liquide est un état moins désordonné qu'un
gaz.
Beaucoup de molécules encastrées entre leurs voisines
ne peuvent que vibrer. Il peut arriver qu'une molécule puisse s'échapper
et échanger sa position avec une autre : c'est l'origine du phénomène
de diffusion, facile à mettre en évidence dans le
cas de deux liquides miscibles.
Pour les liquides, il ny a pas de résultats (ni de théories)
simples
La viscosité des liquides
Quand un liquide n'est pas en équilibre au fond d'un récipient, il coule. Les molécules glissent les unes sur les autres, elles sont maintenues en une masse compacte de volume constant par les forces de cohésion interne qui n'empêchent pas la forme extérieure du liquide de se modifier. Si le glissement est facile, le liquide est fluide et la viscosité est faible. Au contraire la viscosité peut être très élevée, le liquide ressemble à un solide mou. La viscosité est très sensible à la température.
Le solide amorphe
Le cas typique est celui du verre qui, à température ordinaire,
a l'apparence d'un solide très dur et indéformable. Lorsqu'on
augmente la température, il fond sans qu'il y ait discontinuité
dans les propriétés physiques (transition progressive), c'est
à dire sans qu'il y ait de changement dans la structure atomique.
C'est un cas rare de solide appelé solide amorphe ou
vitreux
où l'état solide est en fait un état liquide (donc
une structure désordonnée) de viscosité très
élevée.
3.1. L'état ordonné : le solide cristallisé
Pour l'immense majorité des corps purs, la transition solide-liquide
est discontinue et se produit à température fixe pour une
pression déterminée : suivant le sens, elle est appelée
fusion
ou solidification.
Il existe aussi une transition discontinue solide-gaz : suivant le
sens, c'est la sublimation ou la condensation.
Les solides (à lexception des solides amorphes) ont des structures
ordonnées. On parle de solide cristallisé avec
des maillages périodiques, des " empilements " réguliers.
Cest le domaine de la cristallographie.
On distingue quatre types de solides cristallisés.
- les solides métalliques constitués dions positifs
simples (atomes ayant perdu 1 ou 2 électrons périphériques)
baignant dans un " gaz " délectrons libres ou quasi-libres.
- les solides ioniques constitués dions positifs et négatifs.
- les solides covalents qui sont isolants si la bande dénergie
interdite est large et semi-conducteurs si elle est moindre.
- les solides moléculaires où les interactions de type
Van der Waals expliquent la cohésion.
Lénergie dun solide est constituée de la somme des énergies
de vibration (autour de leur position déquilibre) des particules
de base (ions, atomes ou molécules) et de la somme des énergies
internes des particules de base.
Si la particule de base est un atome ou un ion simple, on obtient, si la température
est suffisante, ![]() .
.
Ce résultat est connu sous le nom de loi de Dulong et Petit
Remarque : entre ordre et désordre
Dans un souci de clarté, nous avons distingué l'état ordonné et l'état désordonné.
En fait, la situation est parfois plus complexe et on parle de structures mal cristallisées ou douées d'un ordre cristallin partiel : dans cette catégorie, on range les hauts polymères (plastiques), beaucoup de substances essentielles à la constitution de nos organes et les cristaux liquides.
4. Changements de phases (détat) des corps purs
Le schéma ci-après donne la nomenclature des divers changements de phase (on dit aussi changements détat ou transitions de phase) entre les états solide, liquide et gazeux.
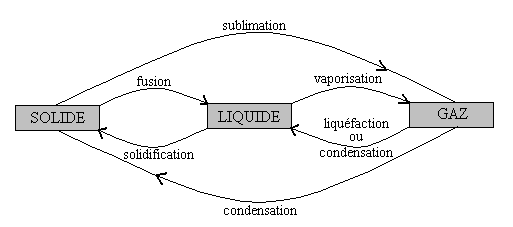
Il convient dêtre précis avec le terme condensation : Il sagit du passage de létat dilué ou raréfié (gazeux ou vapeur) à un état condensé (liquide ou solide). Pour être clair, on devrait préciser, condensation à létat liquide ou condensation à létat solide ; souvent pour le passage à létat liquide, on emploie lexpression liquéfaction lorsquil sagit dun gaz et lexpression condensation lorsquil sagit dune vapeur.
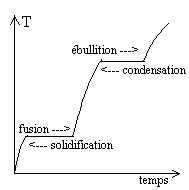 |
Lexpérience courante de transitions
de phase consiste à fournir de lénergie thermique à
un solide à pression constante.
Sa température augmente, puis il fond (la température reste constante durant la fusion). Lorsque le corps est entièrement liquide, sa température recommence à augmenter, puis il bout (sa température reste constante durant lébullition). Lorsque le corps est gazeux, sa température recommence à augmenter. Par refroidissement, on peut faire lexpérience inverse, en passant par les stades de condensation et de solidification. |
Etude expérimentale, courbe de saturation
Aux températures inférieures à la température critique
TC , la compression isotherme dun
gaz provoque sa liquéfaction, cependant nous devons nous situer à
des pressions supérieures à la pression pt
du point triple où coexistent les états
solide, liquide et gazeux.
Si, à partir dune pression faible où létat est gazeux,
on veut réduire le volume, il faut augmenter la pression (un gaz est
facilement compressible). A un certain niveau de pression que nous nommons pression
de vapeur saturante pV , il apparaît
une première goutte de liquide facile à distinguer et, à
partir de là, on peut réduire le volume sans augmenter la pression.
On constate que, dans lenceinte, il y a de plus en plus de liquide. Lorsquil
ny a plus que du liquide, il faut exercer des augmentations de pression très
fortes pour réduire le volume (un liquide est, en première approximation,
incompressible).
Il est possible de faire le processus expérimental inverse en réduisant
les pressions. On part de létat liquide compressé. Lorsquon
atteint la pression de vapeur saturante, il apparaît dans l enceinte
la première bulle de gaz (le liquide se met à bouillir). Tant
que le liquide est en ébullition, la pression reste constante, le volume
augmente et il y a de plus en plus de gaz. Lorsquil ny a plus que du gaz,
il faut réduire la pression pour augmenter le volume.
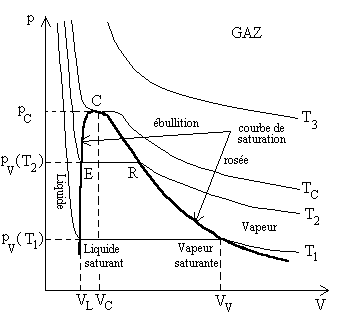 |
Les faits expérimentaux sont repré-sentés
pour différents niveaux de température tels que :
Lensemble des points E est lensemble des points où apparaît la première bulle gazeuse : ils forment la courbe débullition. Lensemble des points R est lensemble des points où apparaît la première goutte de liquide : ils forment la courbe de rosée. La courbe de saturation est formée de la courbe débullition et de la courbe de rosée. Le point C, point supérieur de la courbe de saturation est appelé le point critique. |
Ainsi pour des températures supérieures à la température TC du point critique, un corps ne peut exister quà létat gazeux.
La courbe de compression isotherme, pour la température critique,
présente au point critique un point dinflexion, la tangente y est
horizontale.
En ce point, le corps pur obéit à son équation détat,
à ![]() et à
et à ![]() .
.
Une interprétation du point critique à partir de léquation
détat de Van der Waals ![]() donne :
donne : ![]()
La mesure des coordonnées du point critique fournit des renseignements
sur laspect microscopique de la matière.
Coordonnées du point critique de quelques corps
Vapeur sèche, vapeur saturante, liquide saturant
| Corps | Tc en K | pc en atm | Corps | Tc en K | pc en atm |
| Air | 132,4 | 36 | Méthane | 190,2 | 45 |
| CO2 | 304,1 | 71 | O2 | 154,2 | 49 |
| N2 | 125,9 | 32 | Propane | 370,6 | 39 |
| Butane | 426,3 | 35 | H2 | 32,1 | 19 |
| He | 5,2 | 2,3 | H2O | 647,3 | 218 |
| Isobutane | 406,8 | 36 |
A une température donnée, le changement détat
liquide-gaz se produit à la pression pV
dite pression de vapeur saturante. Cette pression est inférieure
à la pression critique et supérieure à celle du point
triple.
La pression pV ne dépend que
de la température ![]() .
.
Un corps à létat gazeux devient liquide par compression
isotherme lorsquil atteint la pression de vapeur saturante. Inversement
un corps liquide devient gazeux par détente isotherme lorsquil
atteint la pression de vapeur saturante.
A des températures supérieures à la température
critique, un corps est toujours gazeux quelque soit la pression.
Rappelons que létat gazeux à des températures
inférieures à la température critique (qui devient
liquide par compression isotherme) est appelé vapeur sèche
doù le vocabulaire changement détat (ou transition de phase)
liquide-vapeur.
Dans le langage courant, on dit le gaz oxygène, le gaz azote
et la vapeur deau.
La vapeur sèche jusquà sa limite où elle atteint
la pression de vapeur saturante (elle est alors appelée vapeur
saturante) obéit aux mêmes équations détat
que les gaz, en particulier à pressions suffisamment faibles léquation
détat des gaz parfaits est adaptée.
Lorsque la pression dun liquide est celle de la vapeur saturante,
il est appelé liquide saturant.
A lintérieur de la courbe de saturation, nous avons, à
la pression de vapeur saturante, un " mélange " de liquide saturant
et de vapeur saturante que nous appelons vapeur humide.
Liquide saturant et vapeur saturante pris seuls obéissent, en un point
donné, aux équations détat du liquide ou de la vapeur
sèche dont ils sont la limite ; par contre, la vapeur humide a
un comportement tout à fait différent et léquation détat
est ![]() .
.
Vaporisation dans le vide
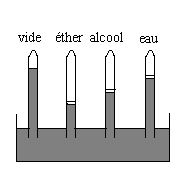 |
Nous introduisons dans les tubes des liquides
différents.
Ils se vaporisent, entièrement si les quantités introduites sont faibles, partiellement si les quantités introduites sont suffisantes (la pression est alors la pression de vapeur saturante). Les dénivellations par rapport au tube de référence sont différentes, la pression de vapeur saturante dépend du corps. A 20 °C pour léther, lalcool et leau, on obtient respectivement des pressions de vapeur saturante de 442, 44 et 17,5 mm de mercure. |
Vaporisation en atmosphère gazeuse
Contrairement à la vaporisation dans le vide qui est " instantanée
", la vaporisation dans un environnement gazeux est un phénomène
lent.
La pression dun mélange gazeux contenant plusieurs constituants
dont certains sont dans des conditions de saturation est égale à
:
![]()
Le premier terme représente la somme des pressions partielles
des divers gaz et vapeurs sèches, le second terme la somme des pressions
des vapeurs saturantes.
Applications du changement détat liquide-vapeur
Létude des machines thermiques nous montrera limportance du
changement détat liquide-vapeur.
Dans ce paragraphe, nous citerons :
- la condensation de leau sur les parois froides et la migration de
lhumidité
- les phénomènes de séchage
- le stockage des fluides
- lexploitation des retards aux changements de phase pour la visualisation
des trajectoires des particules, retard à la condensation dans le
cas des chambres de Wilson, retard à lébullition pour les
chambres à bulle.
4.4. Equilibre liquide-solide, équilibre solide-gaz
Les phénomènes de transitions de phase liquide-solide
et solide-gaz sont analogues à ceux que nous avons étudiés
pour la transition liquide-vapeur.
Cependant, pour ces équilibres, il ny a pas de point critique.
Le phénomène de fusion est courant, celui de sublimation
moins car la pression doit être inférieure à celle
du point triple.
Dans les conditions ordinaires, nous citerons,
- lodeur des cristaux de naphtalène
- dans un tube à essais modérément chauffé,
on observe la coexistence de cristaux diode et de vapeurs violettes diode.
Courbes de sublimation, de fusion, de vaporisation. Point triple
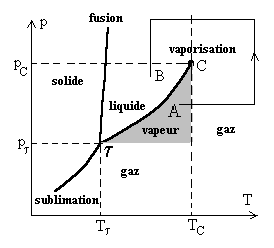 |
Dans un diagramme T, p traçons
les trois courbes déquilibre solide-gaz ou courbe de sublimation,
liquide-vapeur ou courbe de vaporisation, liquide-solide ou courbe de fusion.
Ces trois courbes se coupent nécessairement en un même point appelé point triple t . Au point triple, il y a coexistence des trois phases solide, liquide et gazeuse. Si nous faisons varier soit la pression, soit la température soient ces deux paramètres à partir du point triple, il y a disparition dune ou deux phases. |
La sublimation ne peut se produire pour des pressions supérieures
à celle du point triple.
La courbe de fusion est toujours très proche de la verticale,
généralement de pente positive. Dans le cas de leau, elle
est négative ce qui explique lexpérience amusante du " regel
de leau ".
La courbe de vaporisation est limitée par le point critique C et le point
triple t. Certaines formules
à caractère empirique plus ou moins marqué donnent de bonnes
représentations de ![]() .
.
Nous citerons, la formule de Dupré ![]() ,
celle de Rankine
,
celle de Rankine ![]() ,
celle de Duperray valable pour leau entre 100 °C et 200 °C
à savoir
,
celle de Duperray valable pour leau entre 100 °C et 200 °C
à savoir ![]() où
la pression est en atmosphères et la température en Celsius.
où
la pression est en atmosphères et la température en Celsius.
Enfin, au risque de nous répéter, nous croyons important
dinsister :
- les phénomènes de transitions de phase sont nettement
marqués visuellement (formation de bulles dans un liquide ou dun
solide, dun liquide à la surface dun solide ou dans une vapeur)
et accompagnés de " discontinuités " des propriétés
physiques pour des températures inférieures à la température
critique,
- si nous considérons la transformation AB représentée
sur la figure ci-dessus, nous passons par les états vapeur, gaz
et liquide sans phénomène visuel fortement marqué.
Il y a continuité de létat gazeux et de létat liquide, ces deux phases appartiennent à un même état appelé fluide et diffèrent par une plus ou moins grande densité des molécules.
Il ne faut donc pas sétonner que les liquides obéissent
à des équations détat de même type que les
gaz.
| La figure ci-contre représente les transitions de phase solide, liquide et gaz dans un diagramme p, V . | 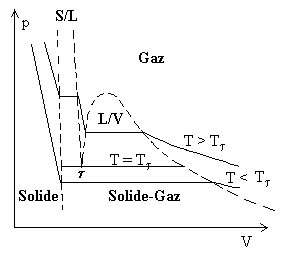 |
Représentation dans un espace à trois dimensions ( p, V, T ) des trois états de la matière d'un corps pur
 |
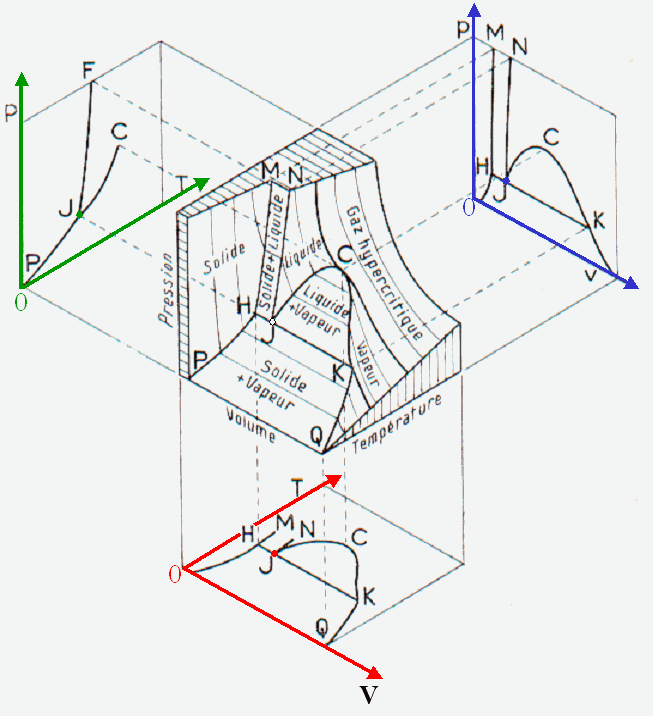 |
Les changements de phase des corps pur saccompagnent déchanges
dénergie considérables.
Cet aspect sera abordé dans le chapitre 4 et complété
dans le chapitre 7
Annexe : la loi de distribution des vitesses de Maxwell
1. Fonction de distribution du vecteur vitesse
Pour un gaz en état déquilibre, posons la question suivante
:
Quelle probabilité existe quune molécule ait ses composantes
de vitesse comprise entre vx et
vx
+ dvx , vy
et vy+ dvy
, vz et vz
+ dvz (repère cartésien
habituel) ?
Cette probabilité d3P dépend
de ![]() (cest
à dire vx , vy
, vz ) et de dvx
, dvy , dvz
. Elle ne dépend pas de la position de la molécule puisque le
gaz est en équilibre.
(cest
à dire vx , vy
, vz ) et de dvx
, dvy , dvz
. Elle ne dépend pas de la position de la molécule puisque le
gaz est en équilibre.
Nous postulons que d3P
est proportionnelle à chacun des intervalles dvx
, dvy , dvz
.
![]()
Il ny a pas de direction privilégiée. Nous pouvons faire
une rotation circulaire des axes cartésiens, la probabilité
sera inchangée (isotropie de lespace).
La fonction ![]() ne
dépend pas du vecteur
ne
dépend pas du vecteur ![]() ,
au mieux elle dépend de lintensité de vitesse v.
,
au mieux elle dépend de lintensité de vitesse v.
![]()
Soit dPx la probabilité
pour une molécule davoir sa composante suivant laxe Ox comprise
entre vx et vx+
dvx .
![]() de même
de même ![]() et
et ![]()
Lisotropie de lespace impose ![]()
La loi des probabilités composées implique :
![]()
Déterminer les fonctions f et jrelève
de la méthodologie mathématique suivante :
![]() avec
avec ![]()
En réorganisant, on obtient : ![]()
Le membre de gauche dépend de v cest à dire de
vx
, vy , vz
, celui de droite uniquement de vx
. Le résultat ne peut être quune constante que nous notons
a .
Il suit que : ![]()
Soit, ![]() et
des expressions identiques pour
et
des expressions identiques pour ![]() .
.
Alors ![]()
Remarquons que a est nécessairement
négatif sinon la probabilité pour une molécule davoir
une composante de vitesse infinie serait infinie ce qui voudrait dire que
toutes les molécules seraient à vitesse infinie et que lénergie
serait infinie !!!
Nous posons a / 2 =-B avec B >
0.
![]()
La condition de normalisation
La probabilité pour une molécule davoir une composante
de vitesse comprise entre -![]() et +
et +![]() ou une intensité
de vitesse comprise entre 0 et +
ou une intensité
de vitesse comprise entre 0 et +![]() est égale à 1 (100% de chances).
est égale à 1 (100% de chances).
Ainsi, par exemple,
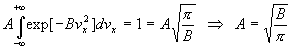
Remarque
Nous avons parlé en terme de probabilité. Un langage équivalent
consiste à chercher, dans une enceinte contenant N molécules,
le nombre dN de molécules ayant certaines caractéristiques,
à savoir par exemple, le nombre de molécules![]() ayant
une composante de vitesse comprise entre vx et vx+
dvx .
ayant
une composante de vitesse comprise entre vx et vx+
dvx .
Ce nombre est égal à : ![]()
2. Fonction de distribution du module de vitesse
Quelle est la probabilité dP(v) pour une molécule
davoir une intensité de vitesse comprise entre v et
v+dv
? ou combien y a til de molécules dN(v) dans une enceinte
contenant N molécules ayant une intensité de vitesse
comprise entre v et v+dv ?
Pour répondre à cette question, nous nous plaçons
dans lespace des vitesses cest à dire un repère
cartésien de coordonnées Ovx
, Ovy , Ovz
.
 |
Les composantes vx
, vy , vz
ne sont pas indépendantes puisque liées par la relation Lextrémité des vecteurs vitesse |
Dans lespace des vitesses, nous délimitons une portion despace comprise
entre la sphère de rayon v + dv et la sphère de rayon v,
de volume égal à ![]() .
.
La probabilité d3P est proportionnelle
à ![]() cest à dire au volume élémentaire dans lespace de vitesse.
Pour obtenir dP(v), nous devons intégrer d3P
à tous les vecteurs vitesse possibles cest à dire ayant leur
extrémité entre les deux sphères.
cest à dire au volume élémentaire dans lespace de vitesse.
Pour obtenir dP(v), nous devons intégrer d3P
à tous les vecteurs vitesse possibles cest à dire ayant leur
extrémité entre les deux sphères.
Cette intégration est particulièrement simple puisque
lintensité v de la vitesse dans cette espace est constante.
On obtient donc : ![]() et aussi, dans une enceinte contenant N molécules, le nombre de
molécules dN(v) ayant un module de vitesse compris entre v
et v + dv : dN(v) = N dP(v).
et aussi, dans une enceinte contenant N molécules, le nombre de
molécules dN(v) ayant un module de vitesse compris entre v
et v + dv : dN(v) = N dP(v).
3. Calcul dune valeur moyenne
La valeur moyenne <G> dune grandeur G est le produit
de G multiplié (pondérée) par la probabilité
dobtenir G intégré à toutes les valeurs possibles
de cette grandeur.
Ainsi, par exemple,
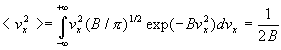
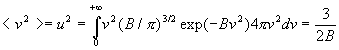
Remarque : ![]()
4. Retour sur la loi de distribution des vitesses
La mesure, pour un gaz parfait monoatomique, de la capacité calorifique à volume constant, conduit à :
Nous disposons de deux expressions de la vitesse quadratique moyenne.
![]()
Les expressions de distribution des vitesses deviennent :

5. Pression dun gaz parfait
On a vu (3.1.2.) linterprétation microscopique de la pression
liée aux chocs des molécules sur une paroi.
Nous considérons le choc des molécules sur un élément
de paroi dS que nous prenons arbitrairement perpendiculaire à
laxe Ox. Le gaz étant en état déquilibre,
toutes les directions sont équivalentes.
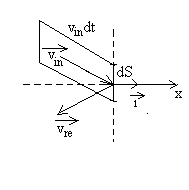 |
Soient Le choc est élastique, la variation de quantité de mouvement pour cette molécule sera |
Combien de molécules de vecteur vitesse ![]() frappent la paroi dS pendant un temps dt ?
frappent la paroi dS pendant un temps dt ?
Elles sont contenues dans un cylindre de base dS, de génératrice
parallèle à ![]() de
longueur
de
longueur ![]() .
Le volume de ce cylindre est égal à
.
Le volume de ce cylindre est égal à ![]() .
.
Le nombre de molécules sera ![]() si N est le nombre de molécules contenues dans lenceinte de volume
V.
si N est le nombre de molécules contenues dans lenceinte de volume
V.
La quantité de mouvement transmise à la paroi par ces molécules
est ![]() que
nous devons intégrer à toutes les molécules venant frapper
la paroi.
que
nous devons intégrer à toutes les molécules venant frapper
la paroi.
Soit 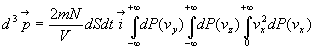
Les deux premières intégrales sont égales à 1, la
troisième à ![]()
En appliquant le principe fondamental de la dynamique, on obtient la
pression à partir de
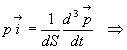
![]()
Retrouver léquation détat des gaz parfaits prouve la validité et la cohérence des hypothèses formulées.
6. Le facteur de Boltzmann
Nous avons introduit la notion de degré de liberté (possibilité
dénergie indépendante) et avons trouvé que, dans le cas
du mouvement de translation suivant un axe, par exemple Ox, le nombre
de molécules ![]() ayant une composante de vitesse vx est
proportionnel à
ayant une composante de vitesse vx est
proportionnel à ![]() .
.
![]() représente
lénergie E suivant ce degré de liberté.
représente
lénergie E suivant ce degré de liberté.
Nous généralisons ce résultat en admettant que le terme ![]() ,
appelé facteur de Boltzmann, caractérise la probabilité
doccupation du niveau dénergie E dans une situation déquilibre
thermique à la température T.
,
appelé facteur de Boltzmann, caractérise la probabilité
doccupation du niveau dénergie E dans une situation déquilibre
thermique à la température T.
Remarques :
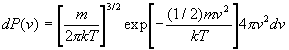 .
.Intégrales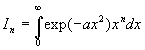 avec
avec ![]()
Nous intégrons par parties en posant ![]()
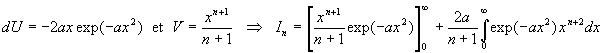
Nous obtenons une relation de récurrence ![]()
A ce stade, nous devons calculer I1 et I0 .
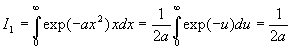
![]()
Le calcul de I0 est plus complexe.
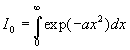 et
et 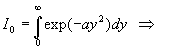
![]()
 |
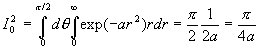
|