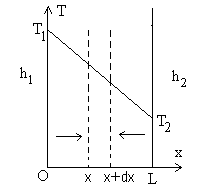Transferts dénergie. Propriétés
de la matière. Bilans énergétiques
Nous limitons nos propos aux variables détat pression, volume,
température et quantité de matière.
On ne saurait être exhaustif dans le domaine des conséquences
des principes. La Thermodynamique est un mode détude applicable
bien sur aux phénomènes thermiques mais pas uniquement. Elle
est applicable aux phénomènes électriques, magnétiques,
chimiques,
elle permet des connaissances spécifiques sur les substances
matérielles.
Nous limitons nos propos à ce dernier aspect et à
des notions sur le transfert thermique.
1. Variables intensives et extensives
Nous considérons un système Shomogène,
sous une seule phase, en équilibre. Les variables détat y
sont uniformes et constantes.
Elles sont liées par une équation détat f(p, V,
T, n ou m) = 0.
Considérons une partie S de S
et notons que dans cette partie pression p et température T
sont inchangées alors que le volume est devenu lV,
la quantité de matière ln ou
lm où l
est compris entre 0 et 1.
Les variables détat ont un caractère différent. On dira
que pression et température sont des variables intensives,
que volume et quantité de matière sont des variables extensives.
Ainsi si nous écrivons léquation détat pour S
sous la forme V = V(T, p, n ou m), elle sécrira pour S
lV= V(T, p, ln ou
lm) cest à dire que la fonction V est une fonction
homogène de degré 1 en n ou en m.
La notation générale que nous utilisons est la suivante : 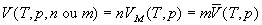
Lindice M indique quune quantité est rapportée
à une mole de matière (on dit molaire), la barre  quelle est rapportée à lunité de masse de matière
(on dit massique).
quelle est rapportée à lunité de masse de matière
(on dit massique).
Nous avons noté le caractère additif des fonctions détat
U et S ; pour un système homogène, sous une phase,
en équilibre ces fonctions seront extensives. Il en sera, de même,
de la fonction détat enthalpie H = U + pV , la quantité
pV produit dune variable intensive par une variable extensive étant
extensive.
2. Travail des forces de pression extérieure
Le piston (paroi mobile en translation) est la technologie la plus
simple pour aborder léchange dénergie par travail provoquant
un changement de létat interne du système. Les études
de technologies liées à des roues mobiles (turbines) sont
plus complexes et font lobjet de cours plus spécialisés
que celui-ci.
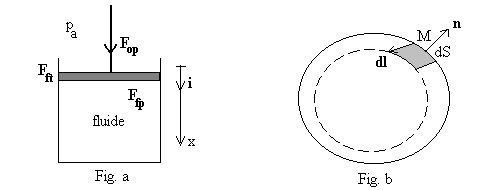
Fig. a - Nous raisonnons sur un piston plan de section S dont
le déplacement est suivant la direction verticale x. Ce cas particulier
nenlève rien à la généralité de nos propos.
Il est soumis à la force de lopérateur extérieur de composante
verticale 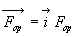 ,
à son poids
,
à son poids 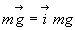 ,
à la pression de lenvironnement
,
à la pression de lenvironnement  ,
à la force tangentielle de frottement de contact
,
à la force tangentielle de frottement de contact 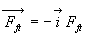 (seule
la projection tangentielle travaille) et à la force
(seule
la projection tangentielle travaille) et à la force 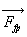 quexerce le système fluide sur le piston (
quexerce le système fluide sur le piston (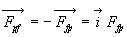 est la force exercée par le piston sur le fluide).
est la force exercée par le piston sur le fluide).
Nous cherchons à connaître le travail dW
échangé par le système fluide du à laction du piston,
soit 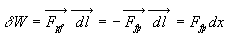 où
où 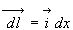 est le vecteur déplacement élémentaire du piston.
est le vecteur déplacement élémentaire du piston.
Lapplication du théorème de lénergie cinétique
au piston permet décrire :
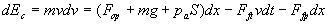
Pour le travail de la force de frottement, il sagit de la vitesse de
glissement du piston sur la paroi du cylindre qui, ici , se confond avec
la vitesse v du piston puisque le mouvement est une translation.
La pression extérieure est définie par la relation 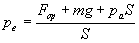 si bien que le travail échangé sécrit
si bien que le travail échangé sécrit 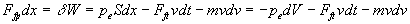 où nous avons introduit
où nous avons introduit 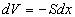 variation de volume du système fluide (pour un déplacement du
piston positif, nous avons une diminution de volume du système ce qui
explique lorigine du signe moins).
variation de volume du système fluide (pour un déplacement du
piston positif, nous avons une diminution de volume du système ce qui
explique lorigine du signe moins).
Lexpression de dW est, même
pour ce cas simplifié, difficilement manipulable car il nous faut
connaître le problème mécanique.
Aussi nous envisageons dW,
- pour un travail des forces de frottement négligeable (le but
étant déchanger du travail avec le système fluide,
les parties mécaniques piston-paroi sont lubrifiées et la
vitesse de glissement est suffisamment faible),
- pour une énergie cinétique négligeable (vitesse
de déplacement du piston suffisamment faible et/ou masse du piston
négligeable),
alors le travail échangé est égal à 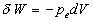 .
.
Fig. b - Dans le cas général, le calcul doit être
fait à chaque élément de surface dS sans faire
dhypothèse sur les directions des forces ou des déplacements.
La pression extérieure 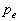 au
point M doit être comprise comme la somme de la pression denvironnement
au
point M doit être comprise comme la somme de la pression denvironnement 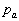 et des projections, suivant la normale à lélément de surface,
des forces extérieures (force dopérateur et poids).
et des projections, suivant la normale à lélément de surface,
des forces extérieures (force dopérateur et poids).
Alors 
(en négligeant le travail des forces de frottement de contact
et lénergie cinétique de lélément de surface)
Remarque : si 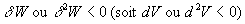 ,
le travail est moteur (le système " pousse " sur le milieu extérieur)
; il est récepteur dans le cas opposé.
,
le travail est moteur (le système " pousse " sur le milieu extérieur)
; il est récepteur dans le cas opposé.
Cas dune pression extérieure uniforme
Dans ce cas là, entre deux instants successifs, nous pouvons intégrer
lexpression de 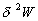 à tous les éléments de surface.
à tous les éléments de surface.
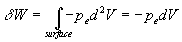 où dV
est la variation de volume du système.
où dV
est la variation de volume du système.
Cas dune transformation quasistatique, dune transformation réversible
Le système est en état déquilibre ou très
voisin dun état déquilibre. Les vitesses de déplacement
ou de glissement sont " nulles ".
La pression du système est uniforme et égale à
p.
En écrivant le principe fondamental de la dynamique sur chaque élément
de surface, on obtient :
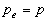 (transformation
réversible ; cette condition traduit aussi la condition déquilibre
mécanique sur une paroi mobile)
(transformation
réversible ; cette condition traduit aussi la condition déquilibre
mécanique sur une paroi mobile)
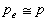 (transformation
quasistatique)
(transformation
quasistatique)
Le travail échangé devient égal à 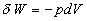
Représentation du travail dans un diagramme V, p
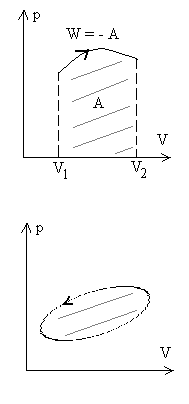 |
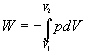 = - aire entre la courbe, les droites
= - aire entre la courbe, les droites 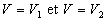 ,
laxe V. ,
laxe V.
Cas dun cycle : W > 0 si le cycle est décrit dans
le sens trigonométrique (ou positif)
W < 0 si le cycle est décrit dans le sens inverse (ou
négatif).
W > 0 le système est récepteur
W < 0 le système est moteur |
Ces schémas montrent que le travail des forces de pression
extérieure dépend des états initial et final, des
états intermédiaires cest à dire de la transformation
amenant de létat initial à létat final (on dit du
chemin suivi).
3. Propriétés de la matière
: détermination des fonctions détat U,
H et
S
3.1. Les caractéristiques thermodynamiques
des corps homogènes
Ces corps obéissent à une équation détat
f(p,
V, T, n ou m) = 0.
Pour un système fermé, la quantité de matière
est fixée et ne doit pas être considérée comme
une variable détat.
Le nombre de variables détat indépendantes pour ces
systèmes est égal à deux et léquation détat
peut être écrite :
V = V(T, p) ou p = p(T, V) ou T = T(p, V) qui
veut dire, par exemple, que le volume V est une fonction des variables
indépendantes T et p.
Les fonctions détat U, H et S seront des fonctions
de deux variables détat indépendantes.
Loutil mathématique nécessaire à la Thermodynamique
repose sur le calcul différentiel. Il nest pas difficile en soi,
il exige de la méthodologie cest à dire un entraînement
obligé.
Le lecteur pourra utiliser lannexe " Eléments de Mathématiques
".
3.1.1. Expressions de la quantité de chaleur d
Q ; coefficients calorimétriques
Le transfert thermique a pour effet de modifier les variables détat
du système (le corps homogène). Dans le cas de variation
de la température, on emploie lexpression chaleur sensible.
Les premier et second principes permettent décrire, par exemple,
en utilisant les variables détat T et
V :
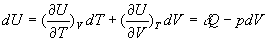 ;
; 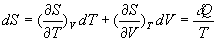
On obtient une forme générale 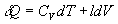 que lon retiendra ainsi que la relation
que lon retiendra ainsi que la relation 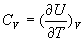
Suivant le couple de variables indépendantes choisies, en appliquant
la même méthode, on obtient trois expressions équivalentes
pour 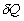 .
.
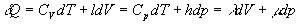 où CV,
Cp, l, h, let
m sont appelés coefficients calorimétriques.
où CV,
Cp, l, h, let
m sont appelés coefficients calorimétriques.
On donne à CVet Cp
le nom propre de, respectivement, capacités calorifiques à
volume constant et à pression constante.
Pour un système homogène, dQ
a
un caractère extensif si bien que, compte tenu du caractère
intensif de T et p, du caractère extensif de V,
les coefficients CV, Cp,
h
et lsont extensifs,
les coefficients l et
m
sont intensifs.
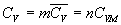 ;
; 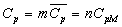 ;
; 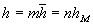 ;
; 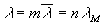
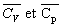 , qui sont respectivement
les capacités calorifiques massiques à volume constant et à
pression constante sont, aussi appelées chaleurs spécifiques à
volume constant et à pression constante.
, qui sont respectivement
les capacités calorifiques massiques à volume constant et à
pression constante sont, aussi appelées chaleurs spécifiques à
volume constant et à pression constante.
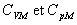 sont les capacités
calorifiques molaires à volume constant et à pression constante.
sont les capacités
calorifiques molaires à volume constant et à pression constante.
Le lecteur pourra établir et retiendra les relations 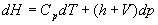 et
et 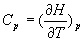
Lunité de mesure de ces différents coefficients calorimétriques
se détermine, sans difficulté, à partir de lanalyse
dimensionnelle (équations aux dimensions) ; ainsi, par exemple,
l se mesure, dans le système MKSA en J/m3.
Relations entre les coefficients calorimétriques
Par exemple, 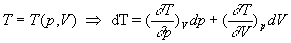
en reportant dans la première expression de dQ
et en identifiant avec la troisième, on obtient les relations 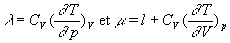
Il est possible de recommencer la procédure autant de fois que
lon peut et de trouver autant de couple de relations entre les coefficients
calorimétriques.
Le lecteur trouvera ci-après les relations utiles entre les
coefficients calorimétriques et nous lui conseillons de savoir les
retrouver.
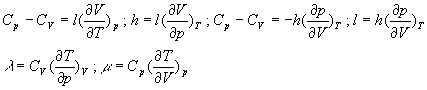
Coefficients thermoélastiques
Ces coefficients (déterminés expérimentalement
et que lon trouve dans les livres de données thermodynamiques)
permettent détablir les équations détat.
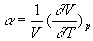 coefficient de
dilatation à pression constante
coefficient de
dilatation à pression constante
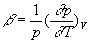 coefficient
daugmentation de pression à volume constant
coefficient
daugmentation de pression à volume constant
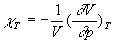 coefficient
de compressibilité isotherme
coefficient
de compressibilité isotherme
La relation mathématique 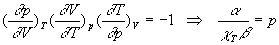
La dérivation de léquation détat donne les coefficients
thermoélastiques, lintégration de deux des coefficients
thermoélastiques fournit léquation détat.
Le lecteur trouvera sans difficulté que, pour un gaz parfait, 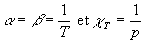
3.1.2. Conséquences mathématiques des premier et second
principes
Variables détat indépendantes T, V
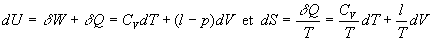
Les deux différentielles totales exactes obéissent à
la relation de Cauchy.
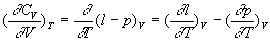
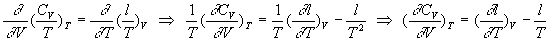
Par identification et dérivation, on obtient : 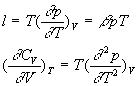
Variables détat indépendantes T, p
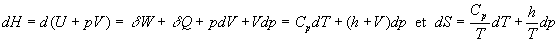
Les deux différentielles totales exactes obéissent à
la relation de Cauchy et on obtient :
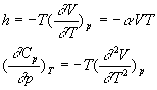
Remarques
- avec les variables T, V on utilise les fonctions énergie
interne U et entropie S ; avec les variables T,
p il convient dutiliser les fonctions enthalpie H et
entropie
S,
- les variables p, V sont dun emploi difficile parce que la
variable T apparaît explicitement dans lexpression de dS,
3.1.3. Détermination des fonctions détat U,
H
et S
3.1.3.1. Tables de données thermodynamiques
En remplaçant l ou h dans la relation entre coefficients
calorimétriques 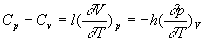 et
en utilisant la relation entre
et
en utilisant la relation entre 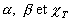 , on obtient :
, on obtient :
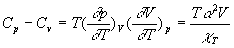
Le coefficient de compressibilité isotherme 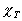 est, pour tous les corps, positif ce qui fait que la capacité calorifique
à pression constante est toujours supérieure à la capacité
calorifique à volume constant (
est, pour tous les corps, positif ce qui fait que la capacité calorifique
à pression constante est toujours supérieure à la capacité
calorifique à volume constant (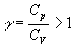 ).
).
Si léquation détat est connue, les coefficients calorimétriques
l et h sont connus, la différence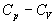 est connue, les coefficients l et m
sont connus si
est connue, les coefficients l et m
sont connus si 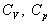 le sont.
le sont.
Du point de vue de la Thermodynamique, la caractérisation
dun corps homogène nécessite la détermination expérimentale
dun coefficient calorimétrique soit CV
soit Cp (en fait Cp
) et de léquation détat (ou des coefficients thermoélastiques).
Dans le chapitre " Description macroscopique de la matière
", nous avons traité de la notion déquations détat
et de leur détermination expérimentale. On trouvera, à
la fin de ce chapitre, une annexe intitulée "Calorimétrie
" dont lobjet est la détermination expérimentale des capacité
calorifiques et des chaleurs latentes.
Les différentielles dU ( ou dH ), dS sont
connues, les fonctions U (ou H ), S aussi à
une constante additive près.
En ce qui concerne lentropie, le troisième principe de la Thermodynamique
(appelé postulat de Nernst-Planck) précise que lentropie
est nulle à 0 K.
Pour ce qui est de lénergie interne ou de lenthalpie, on définit
un état de référence appelé état standard
qui pour les corps purs correspond à la pression standard 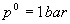 ,
la température étant à préciser (souvent on utilise
la température 25 °C).
,
la température étant à préciser (souvent on utilise
la température 25 °C).
Tout ceci permet de constituer ce quil est convenu dappeler les tables
thermodynamiques.
On est amené à former, suivant les transformations étudiées,
dautres fonctions détat appelées potentiels thermodynamiques.
Nous citerons : lénergie libre 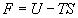 ,
lenthalpie libre
,
lenthalpie libre 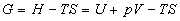 ainsi que les potentiels thermodynamiques généralisés utiles
pour traiter des phénomènes électriques ou magnétiques.
ainsi que les potentiels thermodynamiques généralisés utiles
pour traiter des phénomènes électriques ou magnétiques.
3.1.3.2. Exemple : le
gaz parfait
La méthode est applicable à toute matière dont
on a déterminé léquation détat et dont on
a des renseignements sur lune des capacités calorifiques.
Coefficients calorimétriques
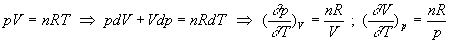
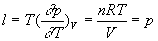
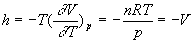
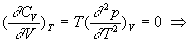
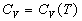 ;
; 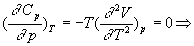
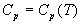
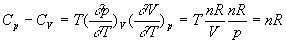 |
Ce résultat est connu sous le nom
de relation de Mayer |
Tous ces résultats théoriques sont conformes aux mesures
présentées dans le tableau de résultats de lannexe
" Détermination expérimentale des caractéristiques
thermodynamiques ".
Lintroduction de 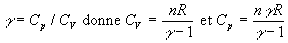
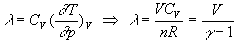 ;
; 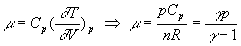
Fonctions détat U, H et S
Energie interne
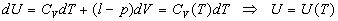
Lénergie interne dun gaz parfait ne dépend que de
la température.
Un gaz dont lénergie interne ne dépend que de la température
obéit à la première loi de Joule.
Ce résultat est validé par lexpérience appelée
détente
de Joule-Gay Lussac.
Pour un gaz monoatomique, 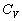 est
constant et égal à
est
constant et égal à 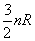 ,
sauf à des températures très voisines de 0 K où varie
rapidement.
,
sauf à des températures très voisines de 0 K où varie
rapidement.
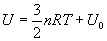
Pour un diatomique, varie
rapidement dans certaines plages de température, en dehors de ces domaines
il est à peu près constant ð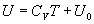
Aux températures ordinaires 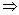
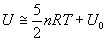
Enthalpie
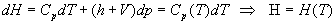
Lenthalpie dun gaz parfait ne dépend que de la température.
Un gaz dont lenthalpie ne dépend que de la température
obéit à la deuxième loi de Joule.
Ce résultat est validé par lexpérience appelée
détente
de Joule-Thomson.
Remarque 1
On sait que les valeurs de H se déduisent directement
des valeurs de U (et inversement) à partir de la définition
à savoir :
H = U + pV = U + nRT.
Or 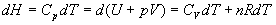 et on
retrouve
et on
retrouve 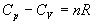
Remarque 2 : un gaz obéissant aux deux lois de Joule est
un gaz parfait
Le gaz obéit à la première loi de Joule : 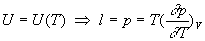
à 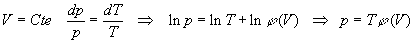
Le gaz obéit à la deuxième loi de Joule : 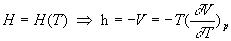
à 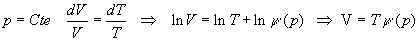
En rapprochant les deux résultats, on obtient 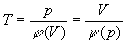
Soit 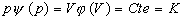 .
En remplaçant y ou j
par sa valeur, il vient pV=KT qui est léquation détat
des gaz parfaits.
.
En remplaçant y ou j
par sa valeur, il vient pV=KT qui est léquation détat
des gaz parfaits.
Un gaz qui obéit aux deux lois de Joule est un gaz parfait.
Entropie
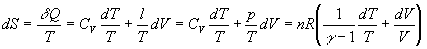
En utilisant les couples de variables indépendantes T, p
ou p, V on obtient des relations similaires.
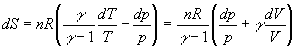
Dans des domaines de température où g
est constant (cest à dire où CV
et Cp sont constants) lintégration
des formules de dS ne présente guère de difficulté.
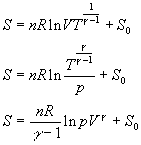
Etude de quelques transformations pour un système fermé
contenant un gaz parfait
Pour toutes les transformations, les capacités calorifiques seront prises
constantes
Transformation isochore (V = Cste)
|
Réalisation : il suffit que les parois
qui délimitent le système soient indéformables et
fixes ; ceci est facilement réalisable pour les systèmes
gazeux, presque impossible pour les liquides ou les solides (dont les
volumes varient peu mais sur lesquels il faudrait exercer des forces très
importantes pour les empêcher de changer de volume)
 ; ; 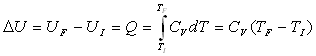
|
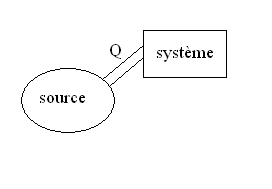
|
Pour une transformation réversible où
la température du système évolue progressivement de 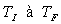 ,
,
on obtient : 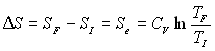 et
et 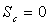

Pour une transformation irréversible où
la source impose " brutalement " une température 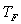 ,
,
on obtient : 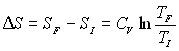 ,
, 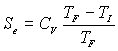 ;
; 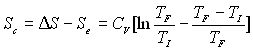
Le lecteur montrera que 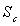 est positif quelques soient
est positif quelques soient 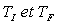 ; il pourra poser
; il pourra poser 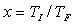 .
.
Transformation isobare (pression extérieure pe
)
|
Réalisation : Le milieu extérieur
impose une pression 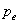 ; dans les états initial ou final, il y a équilibre entre
des pressions entre le milieu extérieur et le système ;
lors de la transformation, le volume du système varie, sa pression
reste constante (transformations quasistatique et réversible) ou
devient non-uniforme (transformation irréversible).
; dans les états initial ou final, il y a équilibre entre
des pressions entre le milieu extérieur et le système ;
lors de la transformation, le volume du système varie, sa pression
reste constante (transformations quasistatique et réversible) ou
devient non-uniforme (transformation irréversible).
|
|
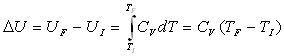 ;
; 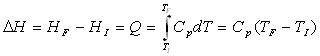 ;
;

Pour une transformation réversible où
la température du système évolue progressivement de ,
on obtient : 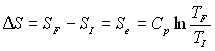 et
et
Pour une transformation irréversible où
la source impose " brutalement " une température ,
on obtient : 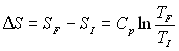 ,
, 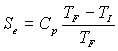 ;
; 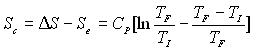
Le lecteur montrera que
est positif quelques soient
; il pourra poser .
Transformation isotherme
Pour une transformation réversible, 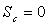 et
et 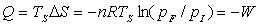
Autre démonstration : dW
= -pdV = Vdp = -dQ
Les variables p et V ne sont pas indépendantes puisque
liées par léquation détat pV=nRTS
On obtient : 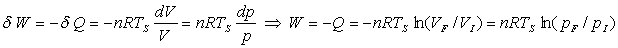
Pour une transformation irréversible où
la pression extérieure 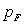 est
imposée " brutalement ",
est
imposée " brutalement ",
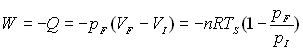
Le calcul de lentropie créée par lirréversibilité
se fait à partir de : 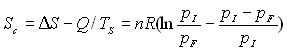 .
.
Le lecteur montrera que
est positif quelques soient  ; il pourra poser
; il pourra poser  .
.
Transformation adiabatique (Q = 0)
|
Réalisation : Le système est
isolé thermiquement de lextérieur cest à dire que
les parois qui délimitent le système sont imperméables
à la chaleur (athermales) ; ceci peut être réalisé
par des parois constitués de matériaux non conducteurs de
la chaleur ou en éliminant le phénomène de convection
et en disposant des écrans anti-rayonnants pour éliminer
les échanges par radiations thermiques.
Le premier principe sécrit DU
= W, le second DS = Sc
.
|
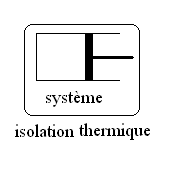
|
Pour une transformation réversible, Sc
est nul donc aussi DS (ð
S est constant).
Une transformation adiabatique réversible est une transformation
isentropique.
En exploitant les formules de lentropie, on obtient une relation suivant
le couple de variables utilisées.
Si g est constant, on retient la relation
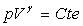 vraie à
chaque instant de la transformation, étant entendu quil est facile de
passer aux autres couples de variables en utilisant léquation détat
des gaz parfaits. Ce résultat est souvent appelé loi de Laplace.
vraie à
chaque instant de la transformation, étant entendu quil est facile de
passer aux autres couples de variables en utilisant léquation détat
des gaz parfaits. Ce résultat est souvent appelé loi de Laplace.
Ainsi, par exemple si le système initialement à pression 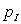 et température
et température 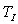 évolue jusqu'à un état final où la pression est
évolue jusqu'à un état final où la pression est
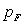 , la température
finale est
, la température
finale est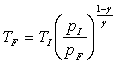 .
.
Le calcul le plus simple de W se fait à partir de 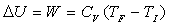 .
On peut aussi faire un calcul direct à partir de
.
On peut aussi faire un calcul direct à partir de 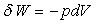 .
.
Pour une transformation irréversible, où,
sur le système initialement à pression
et température ,
on impose " brutalement " une pression extérieure constante
, on calcule W
par deux méthodes,
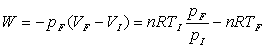 (application
directe de la formule de base)
(application
directe de la formule de base)
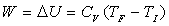
Nous disposons de deux relations indépendantes, de léquation
détat des gaz parfaits dans les états initial et final :
le problème thermodynamique est alors entièrement défini.
En les égalant et en organisant, on obtient 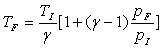
3.2. Etude thermodynamique des changements de phase
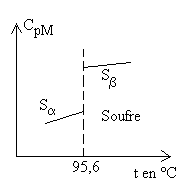
3.2.1. Chaleur latente de changement de phase (détat) dun
corps pur
Lorsque le transfert thermique produit un changement de phase, on emploie
lexpression chaleur latente.
Cette transition de phase seffectue à température et
pression constantes.
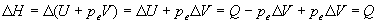
Dans une transformation à pression constante, la quantité
de chaleur échangée est égale à la variation
denthalpie. Ce résultat est important car la variation denthalpie
ne dépend pas de la transformation elle-même.
Définition
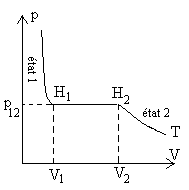 |
On appelle chaleur latente (massique ou molaire)
de changement détat dun corps pur à la température
T
la variation denthalpie (de lunité de masse ou dune mole) de
ce corps passant dun état (solide, liquide ou gazeux) à
un autre état. |
La notation habituelle pour les chaleurs latentes est L. Ainsi 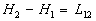 , la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
, la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
Les chaleurs latentes de fusion (transition solide à
liquide), de vaporisation (transition liquide à
vapeur) et de sublimation (transition solide à
gaz) sont positives cest à dire quil faut fournir de la chaleur
pour faire fondre un solide, vaporiser un liquide ou sublimer un solide.
Les chaleurs latentes de solidification, de condensation à létat
liquide et de condensation à létat solide sont respectivement
opposées à celles de fusion, de vaporisation et de sublimation.
Remarque
Les chaleurs latentes sont très importantes. Nous ne donnons
quun seul exemple : pour élever 1 kg deau liquide de 0
°C
à 100 °C, il faut 418
kJ ; pour transformer 1
kg
deau liquide à 100 °C en 1 kg de vapeur à
la même température, il faut 2253 kJ soit environ 4
fois plus.
On trouvera dans lannexe " Calorimétrie " un certain
nombre de résultats expérimentaux et de méthodes de
mesure des chaleurs latentes.
3.2.2. Etude énergétique des vapeurs saturantes. Relations
de Clapeyron
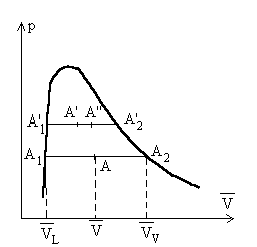 |
Nous raisonnons sur une unité de masse
de corps pur et considérons les deux paliers de liquéfaction
A1A2 et
A1A2
à température T et T + dT.
On appelle x le titre de vapeur saturante défini comme
étant la proportion en masse de vapeur dans le mélange liquide-vapeur.
Pour lunité de masse de corps, x est la masse de vapeur
et 1 - x la masse de liquide.
Soit 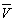 le volume du mélange liquide-vapeur dans létat A.
le volume du mélange liquide-vapeur dans létat A.
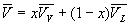 ; ;  sont des fonctions de T suivant la courbe de rosée et la courbe
débullition.
sont des fonctions de T suivant la courbe de rosée et la courbe
débullition. |
La pression de vapeur saturante étant une fonction de T,
les variables détat indépendantes pour le mélange
liquide-vapeur sont x et T.
Etude énergétique
La quantité de chaleur Q échangée pour passer de
A1 à A suivant le palier de liquéfaction
est : 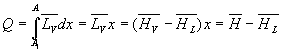
Lénergie interne en A sera : 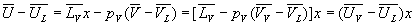
Lentropie en A sera : 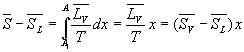
On remarquera lanalogie des formules donnant le titre en fonction
des volumes, des enthalpies, des énergies internes et des entropies.
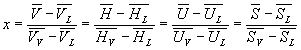
Capacités calorifiques massiques de vapeur saturante et de
liquide saturant
Soit dQ la quantité de chaleur
échangée dans la transformation allant de A2
à A2 suivant la courbe de rosée.
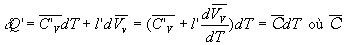 est la capacité
calorifique massique de la vapeur saturante.
est la capacité
calorifique massique de la vapeur saturante.
De même, suivant la courbe débullition, on définit la capacité
calorifique massique du liquide saturant 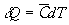
La capacité calorifique du liquide saturant est très
proche (sauf au voisinage du point critique) de la capacité calorifique
du liquide à volume constant.
La capacité calorifique de la vapeur saturante est inférieure
et très différente de celle de la capacité calorifique
à volume constant de la vapeur sèche. Elle est assez souvent
négative, ce qui veut dire que, pour diminuer la température
dune vapeur saturante, il faut lui fournir de la chaleur.
Relations de Clapeyron
Pour lunité de masse de corps, nous considérons la transformation
de létat A(x, T) à létat A(x+dx, T+dT)
en utilisant létat intermédiaire A(x, T+dT).
La quantité de chaleur échangée pour passer de létat
A à létat A est : 
La variation dentropie entre A et A sera : 
La variation denthalpie entre A et A sera : 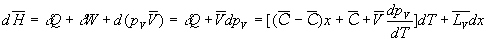
En écrivant la relation de Cauchy sur les deux formes différentielles
qui sont des différentielles totales exactes et en tenant compte de la
relation 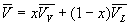 ,
on obtient sans difficulté particulière les très importantes
relations de Clapeyron :
,
on obtient sans difficulté particulière les très importantes
relations de Clapeyron :
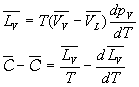
Nous nous sommes appuyés sur léquilibre liquide-vapeur.
Pour
les équilibres liquide-solide et solide-gaz, nous pouvons faire
des raisonnements analogues (en particulier pour les relations de Clapeyron).
Il existe plusieurs démonstrations des relations de Clapeyron,
celle (non présentée) faisant intervenir la fonction détat
enthalpie libre et la notion de potentiel chimique nous parait la plus
intéressante.
" Justification " des formules de Dupré et Rankine
Loin du point critique, 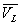 est très inférieur à
est très inférieur à 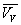 (pour leau
(pour leau 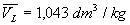 et
et 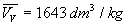 à
100 °C ; la température critique est 374,2 °C),
à
100 °C ; la température critique est 374,2 °C), 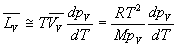 si
on admet que la vapeur sèche jusquà sa limite saturante obéit
à léquation détat des gaz parfaits.
si
on admet que la vapeur sèche jusquà sa limite saturante obéit
à léquation détat des gaz parfaits.
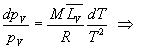 formule de Dupré
si
formule de Dupré
si 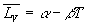 pour
lintervalle de température ou formule de Rankine si
pour
lintervalle de température ou formule de Rankine si 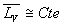 .
.
4. Notions de transferts thermiques
(dénergie calorifique)
On appelle échangeur de chaleur le milieu solide qui sépare
le système de la source de chaleur. Une paroi de faible épaisseur,
de grandes dimensions transversales, est la représentation la plus
simple dun échangeur de chaleur.
4.1. Conduction de la chaleur
La non-uniformité de la température dans un solide entraîne
un
transfert dénergie dun point à un autre. Ce transfert qui
se produit sans transport macroscopique de matière est appelé
" Conduction de la Chaleur ".
Dans les systèmes solides, seul ce mode de transfert est possible. Il
est régi par la loi de Fourier. Dans les fluides, il peut être
secondaire par rapport au transfert par convection.
Nous nous limitons à des solides homogènes et isotropes où,
en chaque point, existe un champ de température 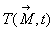
Loi de Fourier
Expérimentalement, si la variation spatiale de température nest
pas trop forte, le vecteur densité de flux de chaleur est égal
à : 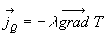
La densité de flux de chaleur j (flux
de chaleur par unité de surface transversale 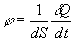 )
dans une direction caractérisée par un vecteur unitaire
)
dans une direction caractérisée par un vecteur unitaire 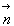 est :
est : 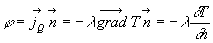
Ce type déquation est appelé équation de diffusion,
lécoulement de chaleur, qui se produit dans le sens des températures
les plus faibles, tend à uniformiser les températures.
Dans le système MKSA, j se
mesure en W.m-2 et
l
en W.m-1.K-1
.
l est appelé conductivité
thermique et traduit laptitude dun matériau à conduire
la chaleur.
Remarque : Lorsque, dans un milieu fluide, existent des gradients
de concentration de matière, il se produit des mouvements de matière
qui tendent à uniformiser la concentration de matière. Ce
phénomène de diffusion de matière est régi
par une loi analogue à celle de Fourier appelée loi de
Fick.
Quelques conductivités thermiques aux températures
ordinaires
| Ordre de grandeur de l
à 20 °C |
W m-1 K-1
|
| Gaz à la pression atmosphèrique |
0,006 - 0,18
|
| Matériaux isolants |
0,025 - 0,25
|
| Liquides non Métalliques |
0,1 - 1,0
|
| Solides non métalliques |
0,025 3
|
| Liquides métalliques |
8,5 85
|
| Alliages métalliques |
10 150
|
| Métaux purs |
20 400
|
| Ag |
418
|
Béton brut |
1,75
|
H2 |
0,18
|
| Cu |
390
|
Verre |
~ 1
|
He |
0,15
|
| Al |
238
|
Plâtre |
0,46
|
Ne |
0,05
|
| Laiton |
120
|
Bois |
0,25 à 0,12
|
O2 |
0,027
|
| Fe |
82
|
Laine de verre |
0,04
|
N2 |
0,026
|
| Pt |
69
|
Polystyrène |
0,04
|
Ar |
0,018
|
| Pb |
35
|
Eau |
0,6
|
CO2 |
0,017
|
| Ti |
20
|
Alcool |
0,17
|
Kr |
0,01
|
| Inox |
14
|
Huile minérale |
0,13
|
|
|
4.2. Echanges de chaleur à la frontière
dun solide
Convection de la chaleur
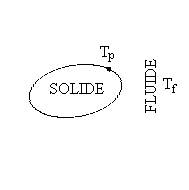 |
Cest le mode de transfert que lon observe
entre un solide et un fluide. Il comprend des phénomènes
de conduction auxquels se superpose un transport de matière, les
molécules du fluide venant se réchauffer ou se refroidir
au contact du solide.
Le transport de matière dans un fluide peut se faire de deux
manières :
-
naturelle les molécules chaudes de masse volumique plus faible
ont tendance à sélever.
|
-
forcée une pompe (ou un ventilateur) provoque le déplacement
du fluide dans une direction.
Létude de ces phénomènes, régis par les équations
de Navier-Stokes, ne peut être abordée sans de sérieuses
connaissances de Mécanique des fluides.
Nous resterons très élémentaire et poserons que
la densité de flux de chaleur échangée par convection
est égale à :
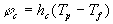 où hc
est le coefficient de convection, Tp la
température à la paroi du solide et Tf
la " température du fluide " au loin si celui-ci est " seul ", de mélange
sil est entre deux parois. hc est exprimée
en W.m-2.K-1.
où hc
est le coefficient de convection, Tp la
température à la paroi du solide et Tf
la " température du fluide " au loin si celui-ci est " seul ", de mélange
sil est entre deux parois. hc est exprimée
en W.m-2.K-1.
hc est de lordre de quelques
unités pour la convection naturelle des gaz, quelques dizaines pour
la convection forcée des gaz, quelques centaines pour la convection
naturelle des liquides et de quelques milliers pour la convection forcée
des liquides.
Rayonnement thermique
La surface dun solide émet un rayonnement électromagnétique
complexe dénergie dautant plus grande que cette surface est à
température plus élevée .
Ce rayonnement se propage dans les milieux transparents. Lorsquil
rencontre un corps, il est totalement absorbé (corps noir), partiellement
absorbé (corps réfléchissants, semi-transparents)
ou pas du tout absorbé (corps parfaitement réfléchissants,
corps transparents). Lénergie absorbée est transformée
en énergie interne.
Le spectre du rayonnement électromagnétique dépend
de la température de surface du corps qui émet, de la nature
du corps et de son état de surface.
Laptitude dun corps à réfléchir, transmettre
ou absorber un rayonnement dépend aussi de ces paramètres.
Un bilan énergétique montre que, pour un corps en équilibre
thermique, son aptitude à absorber un rayonnement est identique à
son aptitude à en émettre. On rend compte de cette aptitude par
un coefficient 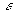 compris entre 0 et 1, appelé émissivité.
compris entre 0 et 1, appelé émissivité.
Dans une direction 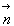 ,
la puissance énergétique spectrale (flux énergétique)
,
la puissance énergétique spectrale (flux énergétique) 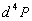 dun rayonnement de longueur donde compris entre
dun rayonnement de longueur donde compris entre 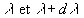 ,
émis (ou absorbé) par un élément de surface
,
émis (ou absorbé) par un élément de surface 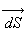 dans un angle solide
dans un angle solide 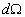 par la relation :
par la relation :
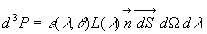 où
où 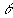 est langle entre
est langle entre  et
et 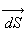 et
et 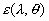 lémissivité spectrale dans la direction qui traduit laptitude
du corps à émettre ou absorber.
lémissivité spectrale dans la direction qui traduit laptitude
du corps à émettre ou absorber.
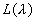 est la luminance
spectrale du corps noir qui est un corps qui absorbe tout rayonnement
[
est la luminance
spectrale du corps noir qui est un corps qui absorbe tout rayonnement
[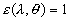 ].
].
Suivant une théorie de Planck introduisant la notion de quantum
dénergie,
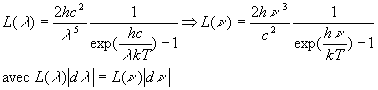

La valeur 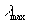 correspondant
à un maximum de la luminance spectrale est donnée par la loi de
Wien, à savoir
correspondant
à un maximum de la luminance spectrale est donnée par la loi de
Wien, à savoir 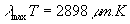
Lémissivité des corps obéissant à la
loi de Lambert ne dépend pas de la direction, par suite
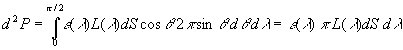
La quantité  est
appelée émittance spectrale
est
appelée émittance spectrale
Pour des corps ternes (dit gris ; cas des peintures non métalliques,
des matériaux servant dans la construction), on peut considérer
que 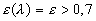 .
.
On en déduit la densité de flux énergétique par
rayonnement : 
La densité de flux dénergie émis par un corps sous forme
de rayonnement électromagnétique par un corps à température
T est égale à 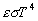 où
où 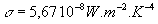 est la constante de Stéfan-Boltzmann.
est la constante de Stéfan-Boltzmann.
Pour des corps métalliques polis (brillants et donc réfléchissants),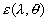 est
spectral et directionnel. Ses valeurs sont de lordre de 0,05 à 0,3.
est
spectral et directionnel. Ses valeurs sont de lordre de 0,05 à 0,3.
Les échanges dénergie par rayonnement entre deux corps
dépendent de leur position relative.
En restant très élémentaire, la densité de flux
de chaleur échangée par rayonnement est : 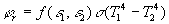
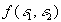 est une fonction
des émissivités des surfaces et de leur position relative.
est une fonction
des émissivités des surfaces et de leur position relative.
-
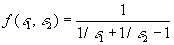 dans le cas
de deux surfaces planes, parallèles, de grandes dimensions,
dans le cas
de deux surfaces planes, parallèles, de grandes dimensions,
-
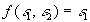 si la surface
démissivité
si la surface
démissivité 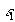 est convexe et entièrement entourée par une surface de très
grande dimension.
est convexe et entièrement entourée par une surface de très
grande dimension.
En remarquant que 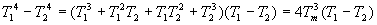 si Tm est une température moyenne
entre T1 et T2,
la densité de flux de chaleur échangée par rayonnement peut
être linéarisée sous la forme
si Tm est une température moyenne
entre T1 et T2,
la densité de flux de chaleur échangée par rayonnement peut
être linéarisée sous la forme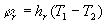 avec
avec 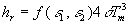 .
.
Cette linéarisation est très utilisée lorsque
les températures T1 et T2
sont voisines car hr , coefficient
de rayonnement, est pratiquement constant.
5. Bilans énergétiques
simples faisant intervenir les transferts thermiques
5.1. Paroi solide inerte déchangeur en régime
permanent
Pour ce type de système, il ny a pas, au cours du temps, de
variation de son énergie totale, ni déchange de travail
avec le milieu extérieur puisque le volume est constant.
Le principe de conservation de lénergie impose quà
chaque instant la chaleur échangée est nulle.
Mathématiquement, on écrit que le flux de chaleur 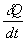 échangé par le système est nul (à chaque instant,
le flux de chaleur
échangé par le système est nul (à chaque instant,
le flux de chaleur 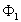 " entrant " est égal au flux de chaleur
" entrant " est égal au flux de chaleur 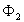 " sortant ", soit
" sortant ", soit 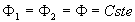 puisque le régime est permanent).
puisque le régime est permanent).
Dans le cas particulier où, dans le bilan énergétique,
seuls interviennent des échanges de chaleur, on emploie lexpression
bilan thermique.
5.1.1. Paroi en forme de " mur "
En thermique, on appelle " mur " un système où les échanges
de chaleur se produisent suivant une direction cartésienne, par
exemple x.
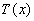 représente
donc le champ de température en régime permanent.
représente
donc le champ de température en régime permanent.
Le bilan thermique consiste à écrire que le flux de chaleur
qui entre est égal à celui qui sort.
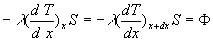
Ainsi  (on trouve
(on trouve 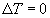 ,
cas particulier, pour un mur inerte en régime permanent, de léquation
de la chaleur).
,
cas particulier, pour un mur inerte en régime permanent, de léquation
de la chaleur).
La résolution, avec les deux conditions aux limites 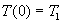 et
et 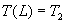 , conduit
à
, conduit
à 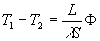 et
et 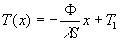
-
 représente
la résistance thermique pour une section transversale S
et
représente
la résistance thermique pour une section transversale S
et 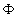 le
flux de chaleur. Nous pouvons faire une analogie avec le domaine électrique
en régime permanent entre différences de température
et différence de potentiel dune part et, dautre part, flux de chaleur
et intensité du courant électrique.
le
flux de chaleur. Nous pouvons faire une analogie avec le domaine électrique
en régime permanent entre différences de température
et différence de potentiel dune part et, dautre part, flux de chaleur
et intensité du courant électrique.
- A la frontière du solide, en
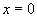 et
et 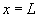 , les
échanges de chaleur se produisent respectivement avec des fluides à
température
, les
échanges de chaleur se produisent respectivement avec des fluides à
température 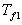 et
et 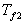 par
lintermédiaire de coefficients déchanges de chaleur
par
lintermédiaire de coefficients déchanges de chaleur 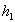 et
et 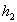 .
.
Le bilan énergétique (thermique) permet décrire
:
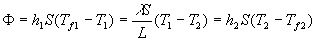
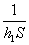 et
et 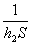 apparaissent comme les résistances thermiques liées au coefficients
et et
apparaissent comme les résistances thermiques liées au coefficients
et et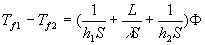
Remarque : utilisation " d'ailettes ou barres " pour augmenter les
échanges de chaleur
On place, aux frontières de la paroi, des solides de forme particulière
appelés " ailettes ou barres ". Laugmentation des échanges
de chaleur (diminution de la résistance thermique) est obtenue par
laugmentation de la surface déchange avec les fluides extérieurs.
Ces solides sont de forme allongée, cest à dire possèdent
des dimensions transversales faibles par rapport à la troisième
dimension.
Le gradient de température a lieu principalement dans le sens
de cette dernière direction. Chaque section transversale est, en
première approximation, à température uniforme ce
qui nest pas contradictoire avec le fait de considérer des échanges
de chaleur suivant ces directions jusquau contact avec le fluide extérieur.
L efficacité 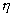 dune ailette est définie par le rapport du flux de chaleur échangé
par lailette au flux de chaleur qui serait échangé si elle nexistait
pas.
dune ailette est définie par le rapport du flux de chaleur échangé
par lailette au flux de chaleur qui serait échangé si elle nexistait
pas.
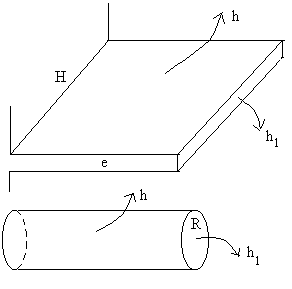
Ailettes à section uniforme (rectangulaire ou circulaire)
Le flux de chaleur évacué par lailette peut être
calculé de deux manières :
- en calculant le flux de chaleur du aux coefficients déchanges
suivant les éléments de surface en contact avec lextérieur,
- en calculant (plus agréable) le flux de chaleur entrant par
conduction dans lailette.
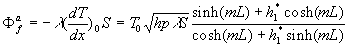
Pour une ailette très longue (semi-infinie), on obtient
:
 (le lecteur
reprendra la résolution avec
(le lecteur
reprendra la résolution avec 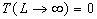 ou fera tendre L vers linfini dans les résultats de lailette
finie ; dans ce dernier cas, il obtiendra la condition
ou fera tendre L vers linfini dans les résultats de lailette
finie ; dans ce dernier cas, il obtiendra la condition 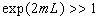 ).
).
Lefficacité de cette ailette est 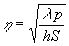
-
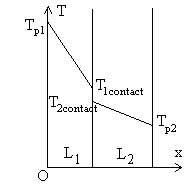
Examinons le cas de deux murs " accolés " (de même section
avec une face commune)
Le problème posé est celui de lécart 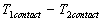 .
.
Lextrapolation jusquau contact du champ de température dans le milieu
1 conduit à la valeur 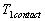 qui est, à priori, différente de
qui est, à priori, différente de 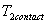 température
dextrapolation jusquau contact dans le milieu 2.
température
dextrapolation jusquau contact dans le milieu 2.
Pour tenir compte de cet écart, on introduit une résistance
thermique supplémentaire appelée résistance thermique
de contact suivant la relation 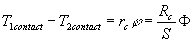
Les valeurs de la résistance de contact sont sensibles
si la conductivité du fluide interstitiel est très différente
de celles constituant les matériaux en contact, donc dans le cas
de matériaux en contact conducteurs.
Si lécart
est négligeable, on dit que le contact est parfait.
On remarque, de plus, que 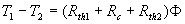
5.1.2. Paroi à symétrie de révolution. Paroi
à symétrie sphérique
Symétrie de révolution autour dun axe
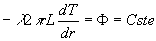 ;
; 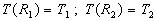 où r est la distance à laxe et L une longueur axiale arbitraire.
où r est la distance à laxe et L une longueur axiale arbitraire.
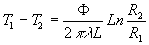 et
et 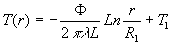 .
.
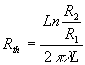 représente
la résistance thermique (
représente
la résistance thermique ( ).
).
Symétrie sphérique
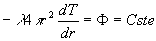 ;
;  où r est la distance au centre de symétrie.
où r est la distance au centre de symétrie.
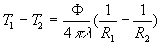 et
et 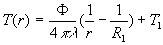
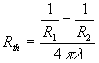 représente
la résistance thermique ().
représente
la résistance thermique ().
5.2. Bilans énergétiques pour un solide
en régime transitoire
Entre deux instants successifs t et t + dt, le premier
principe de la Thermodynamique pour un système peut être écrit
:
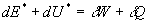 où
où
-
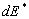 doit être
compris comme la transformation au sein du milieu dénergie potentielle,
dénergie chimique ou nucléaire en énergie calorifique
; il sagit de leffet Joule ou de lénergie calorifique résultat
dune réaction exothermique ou endothermique (nous appelons sources
de chaleur internes ce type dénergie)
doit être
compris comme la transformation au sein du milieu dénergie potentielle,
dénergie chimique ou nucléaire en énergie calorifique
; il sagit de leffet Joule ou de lénergie calorifique résultat
dune réaction exothermique ou endothermique (nous appelons sources
de chaleur internes ce type dénergie)
-
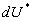 est la variation
dénergie interne résultant de variations des variables détat
est la variation
dénergie interne résultant de variations des variables détat
-
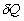 représente
les échanges de chaleur aux frontières du système
représente
les échanges de chaleur aux frontières du système
-
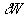 représente
les échanges de travail aux frontières du système.
représente
les échanges de travail aux frontières du système.
Le système solide subit une transformation à pression 
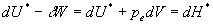 variation denthalpie
résultant de variation dans le temps du champ de température.
variation denthalpie
résultant de variation dans le temps du champ de température.
5.2.1. Cas où la température du solide est uniforme
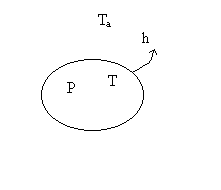 |
On considère un milieu de volume V,
de surface S, de capacité calorifique 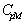 ,
siège dune dissipation dénergie de puissance ,
siège dune dissipation dénergie de puissance 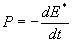 . .
La température extérieure 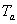 est constante, celle du milieu
est constante, celle du milieu 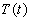 est uniforme . On appelle h le coefficient déchange traduisant
les échanges de chaleur par convection et/ou rayonnement.
est uniforme . On appelle h le coefficient déchange traduisant
les échanges de chaleur par convection et/ou rayonnement. |
Le bilan thermique conduit à 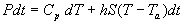 ,
soit
,
soit 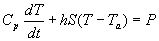
En régime permanent, on obtient 
5.2.2. Equation de la chaleur
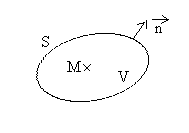
On considère un élément
du milieu quelconque, suffisamment petit pour être homogène,
de volume V, limité par une surface S.
-
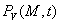 est
la puissance calorifique volumique des sources internes, si bien que est
la puissance calorifique volumique des sources internes, si bien que 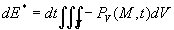
-
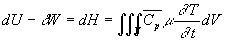 où où 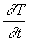 est lélévation de température par unité
de temps,
est lélévation de température par unité
de temps,
|
 |
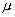 et
et 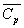 respectivement la masse volumique et la capacité calorifique massique
à pression constante du milieu (on remarquera que, dans le cas dune
évolution à volume constant, il conviendrait de remplacer la capacité
calorifique à pression constante par celle à volume constant et
que la différence nest significative que dans le cas des gaz).
respectivement la masse volumique et la capacité calorifique massique
à pression constante du milieu (on remarquera que, dans le cas dune
évolution à volume constant, il conviendrait de remplacer la capacité
calorifique à pression constante par celle à volume constant et
que la différence nest significative que dans le cas des gaz).
- Les échanges de chaleur aux frontières du milieu sexpriment,
compte tenu de lorientation de la normale
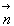 ,
par la relation
,
par la relation 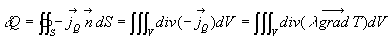
Lapplication du premier principe de la Thermodynamique conduit à léquation
de la chaleur,
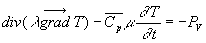
Pour des conductivités thermiques indépendantes de la température
et des milieux isotropes, on obtient :
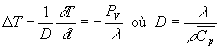 est la diffusivité
thermique (m2.s-1).
est la diffusivité
thermique (m2.s-1).
5.3. Bilan énergétique pour un fluide
en écoulement permanent
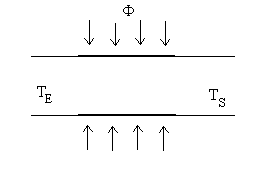 |
Lécoulement permanent dun fluide seffectue
à pression constante.
Il reçoit un flux de chaleur  si bien que sa température évolue dune valeur amont (à
lentrée)
si bien que sa température évolue dune valeur amont (à
lentrée) 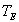 à une valeur aval (à la sortie)
à une valeur aval (à la sortie) 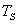 . .
Une quantité de masse dm de fluide échange la quantité
de chaleur 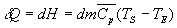 si on néglige, pour le fluide, toute variation dénergie cinétique
et potentielle
si on néglige, pour le fluide, toute variation dénergie cinétique
et potentielle |
Par unité de temps, on écrit 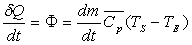 où
est le flux de chaleur échangé par le fluide.
où
est le flux de chaleur échangé par le fluide.
Annexe : Calorimétrie
La calorimétrie a pour objet la mesure des capacités calorifiques
et des chaleurs latentes de changement détat.
1. Mesure des capacités calorifiques
1.1. Méthodes de mesure pour les solides, les liquides
En réalisant une transformation isobare, on détermine expérimentalement
Cp . On calcule CV
à partir de 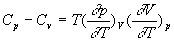 .
.
En fait les écarts relatifs 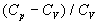 restent faibles, 5% pour laluminium, 4% pour lor, 3% pour le cuivre et largent,
... de lordre de grandeur des erreurs de mesures.
restent faibles, 5% pour laluminium, 4% pour lor, 3% pour le cuivre et largent,
... de lordre de grandeur des erreurs de mesures.
La méthode des mélanges
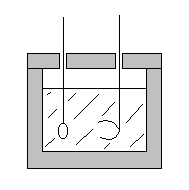 |
Un vase est rendu pratiquement adiabatique par
une enveloppe isolante (ou un système disolation complexe). Un tel
système est appelé calorimètre adiabatique.
On y introduit une masse deau de capacité calorifique massique à
pression constante 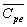 . Soit
. Soit 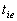 la température initiale de leau et du vase supposés en équilibre
thermique. Un corps solide de masse M est extrait dune étuve
où il était en équilibre à température
la température initiale de leau et du vase supposés en équilibre
thermique. Un corps solide de masse M est extrait dune étuve
où il était en équilibre à température 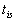 (en général, on choisit
(en général, on choisit 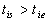 ).
On plonge le solide de capacité calorifique massique à pression
constante ).
On plonge le solide de capacité calorifique massique à pression
constante 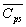 dans le calorimètre le plus rapidement possible et on referme le
couvercle.
dans le calorimètre le plus rapidement possible et on referme le
couvercle. |
On agite afin datteindre un équilibre à température finale 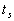 au plus vite.
au plus vite.
Soit m la masse deau qui échangerait
dun point de vue thermique comme le vase et les accessoires (agitateur,
capteur de température).
La transformation est irréversible, isobare à la pression 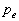 de
lenvironnement (le plus souvent pression atmosphérique).
de
lenvironnement (le plus souvent pression atmosphérique).
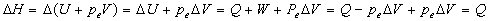
Pour le système, corps solide + calorimètre, la quantité
de chaleur, donc la variation denthalpie, est nulle.
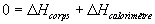
Lenthalpie est une fonction détat, sa variation ne dépend que
des états initial et final.
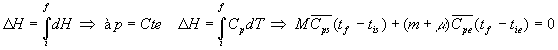
soit 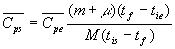
Cette méthode nécessite la mesure préalable de
m
qui pourra être faite par la méthode électrique que
nous allons voir ou en faisant une expérience préalable avec
une masse m deau à température connue à la place
du solide.
La méthode électrique
Particulièrement adaptée pour les liquides, elle consiste
à chauffer le liquide enfermé dans le calorimètre
adiabatique à laide dune résistance électrique.
La connaissance de lénergie électrique fournie et de lélévation
de température permet la mesure de la capacité calorifique.
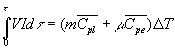
V : tension aux bornes de la résistance ; I : courant électrique
; m : masse du liquide ; 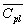 capacité calorifique massique à pression constante du liquide
; m : valeur en eau du calorimètre; :
capacité calorifique massique à pression constante de leau ;
DT : élévation de température.
capacité calorifique massique à pression constante du liquide
; m : valeur en eau du calorimètre; :
capacité calorifique massique à pression constante de leau ;
DT : élévation de température.
Remarques :
- un calorimètre nest jamais parfaitement adiabatique (des
corrections sont nécessaires faisant appel aux transferts thermiques),
- dans les microcalorimètres, on nempêche pas les pertes
thermiques, on les favorise mais on en tient compte avec précision,
- il sagit, sur lintervalle de température, des valeurs moyennes
des capacités calorifiques doù lintérêt de
travailler pour des écarts de température les plus faibles
possibles,
- en Transferts Thermiques (Conduction de la chaleur), dautres méthodes
existent.
1.2. Méthodes de mesure pour les gaz
Ces mesures sont difficiles en raison de la faiblesse des masses volumiques
des gaz et de lobligation de les enfermer dans des calorimètres
dont la capacité calorifique est souvent plus importante que celle
du gaz.
On mesure 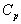 et lon déduit
et lon déduit 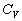 soit de la formule
soit de la formule 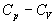 ,
soit de la mesure de
,
soit de la mesure de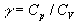 .
.
Une méthode de mesure de 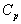 dun gaz est connue sous le nom de méthode du courant stationnaire.
Elle est adaptée aux liquides.
dun gaz est connue sous le nom de méthode du courant stationnaire.
Elle est adaptée aux liquides.
Une méthode de mesure de g
est connue sous le nom dexpérience de Clément et Desormes.
Un autre processus de mesure de g porte le
nom de formule de Reech.
Nous avons introduit le coefficient de compressibilité isotherme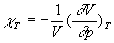 ,
nous introduisons celui de compressibilité adiabatique
,
nous introduisons celui de compressibilité adiabatique 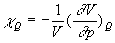 et remarquons que
et remarquons que 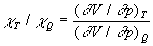 en un même état.
en un même état.

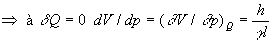
Ainsi, 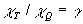 .
.
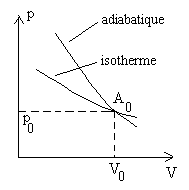 |
Traçons dans un diagramme V, p (diagramme
de Clapeyron) lisotherme passant par le point A0
de coordonnées (p0 , V0).
Les dérivées 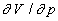 représentent linverse de la pente de ces courbes et on obtient :
représentent linverse de la pente de ces courbes et on obtient :
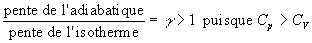
En un même point, la pente de ladiabatique, dans un diagramme
V,
p, est toujours plus accentuée que celle de lisotherme. |
1.3. Résultats
Cas des solides
Le tableau ci-dessous donne la capacité calorifique massique
et molaire à pression constante pour différents corps simples
à température et pression ordinaires ainsi que les masses
molaires M.
| |
Be
|
B
|
C
|
Al
|
P
|
Fe
|
Cu
|
Ag
|
Sb
|
Pb
|
| |
|
|
diamant
|
|
blanc
|
|
|
|
|
|
| M en g |
9
|
10,8
|
12
|
27
|
31
|
55,8
|
63,5
|
108
|
122
|
207
|
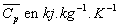 |
1,63
|
1,0
|
0,50
|
0,88
|
0,79
|
0,46
|
0,39
|
0,23
|
0,21
|
0,13
|
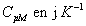 |
14,7
|
10,8
|
6,0
|
23,7
|
24,6
|
25,6
|
24,7
|
24,8
|
25,5
|
26,8
|
On constate, sauf pour les éléments légers tels que Be, B
et C, que la capacité calorifique molaire est à peu près  (loi de Dulong et Petit, voir chapitre 2)
(loi de Dulong et Petit, voir chapitre 2)
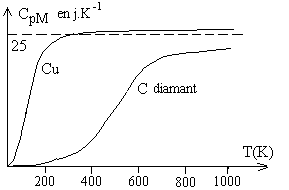 |
En fait 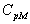 varie avec la température (fonction croissante) et, pour tous les
corps, tend vers 0 si la température tend vers 0.
varie avec la température (fonction croissante) et, pour tous les
corps, tend vers 0 si la température tend vers 0.
Si la température est suffisante, 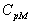 atteint une valeur limite autour de 3R. Pour les éléments
légers aux températures ordinaires, la valeur limite nest
pas atteinte.
atteint une valeur limite autour de 3R. Pour les éléments
légers aux températures ordinaires, la valeur limite nest
pas atteinte. |
Cas des liquides
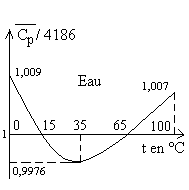 |
Il ny a pas de loi simple. Pour de nombreux liquides, on trouve un 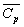 de lordre de 2000 j.kg-1.K-1
exception faite de leau dont la valeur est double. Cette propriété,
jointe au fait que cest un produit bon marché, fait de leau un
fluide caloporteur (porteur de chaleur) de grand intérêt, très
employé dans les circuits de refroidissement des moteurs, dans le
chauffage central, dans les machines thermiques, ...
de lordre de 2000 j.kg-1.K-1
exception faite de leau dont la valeur est double. Cette propriété,
jointe au fait que cest un produit bon marché, fait de leau un
fluide caloporteur (porteur de chaleur) de grand intérêt, très
employé dans les circuits de refroidissement des moteurs, dans le
chauffage central, dans les machines thermiques, ... |
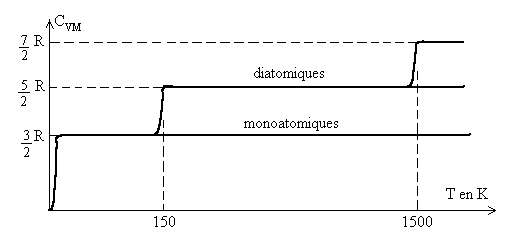
Cas des gaz
Le tableau ci-après et les courbes représentées
rassemblent les principaux résultats.
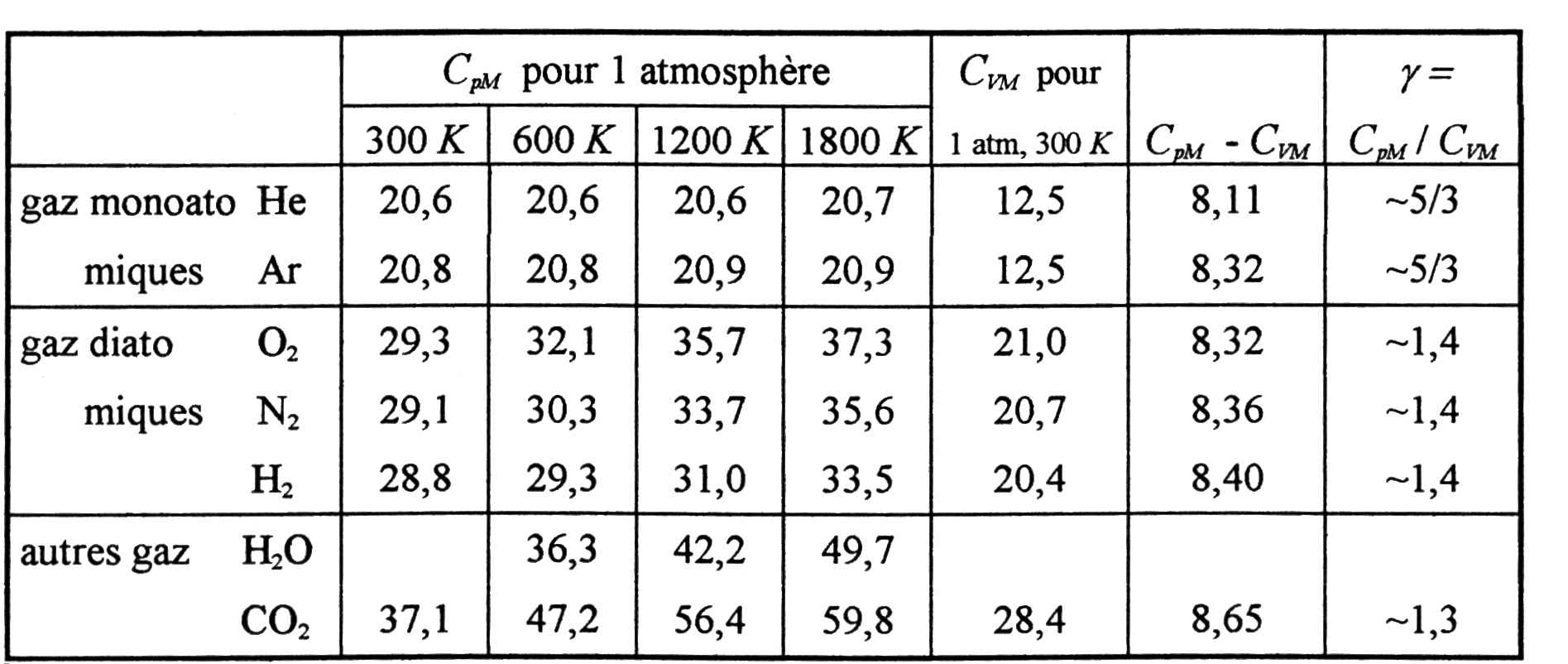
Commentaires : Les résultats sont donnés pour une
pression atmosphérique normale pour laquelle, en première
approximation, les gaz réels ont un comportement de gaz parfait.
Les courbes traduisent les évolutions de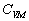 avec la température, des différences apparaissent avec latomicité.
avec la température, des différences apparaissent avec latomicité.
La valeur de 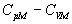 est égale à R quelque soit le gaz.
est égale à R quelque soit le gaz.
A la température T = 0 K, la valeur de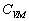 est nulle. Elle évolue très rapidement (entre 0 et 1K)
vers la valeur
est nulle. Elle évolue très rapidement (entre 0 et 1K)
vers la valeur  quel que soit le gaz.
quel que soit le gaz.
Les gaz monoatomiques gardent cette dernière valeur quelque
soit la température.
Les autres gaz subissent deux évolutions pour ,
lune autour de 150 K, lautre autour de 1500 K.
Pour les diatomiques,
atteint la valeur 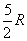 ,
puis
,
puis 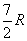 . Pour
une atomicité supérieure, les résultats sont plus complexes.
Ils dépendent de la forme de la molécule et de son atomicité.
On atteint dabord
. Pour
une atomicité supérieure, les résultats sont plus complexes.
Ils dépendent de la forme de la molécule et de son atomicité.
On atteint dabord 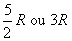 ,
puis une valeur plus élevée.
,
puis une valeur plus élevée.
2. Chaleurs latentes de changement de phase
2.1. Définition
|
A pression constante, la variation denthalpie
est égale à la quantité de chaleur échangée
dans la transformation.
On appelle chaleur latente (massique ou molaire) de changement de
phase dun corps pur à la température T la variation
denthalpie (de lunité de masse ou dune mole) de ce corps passant
dun état (solide, liquide ou gazeux) à un autre état. |
La notation habituelle pour les chaleurs latentes est L. Ainsi 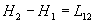 , la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
, la variation denthalpie pour aller de létat 1 à létat
2 est égale à la quantité de chaleur échangée
sur lisobare.
2.2. Mesure des chaleurs latentes
Chaleur latente de fusion
On utilise un calorimètre adiabatique maintenu à une température
constante
légèrement supérieure à la température de
fusion 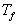 du
corps à étudier. On y introduit une masse m de ce corps
à une température
du
corps à étudier. On y introduit une masse m de ce corps
à une température 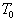 légèrement inférieure à
(le corps est donc en phase solide) ; on maintient la température du
calorimètre constante à laide dune résistance immergée
dans le calorimètre et parcourue par un courant électrique réglable.
légèrement inférieure à
(le corps est donc en phase solide) ; on maintient la température du
calorimètre constante à laide dune résistance immergée
dans le calorimètre et parcourue par un courant électrique réglable.
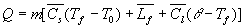 , si Q
est la quantité de chaleur fournie par effet Joule,
, si Q
est la quantité de chaleur fournie par effet Joule,  les capacités calorifiques massiques du corps à létat
solide et liquide et
les capacités calorifiques massiques du corps à létat
solide et liquide et 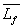 la chaleur latente massique de fusion du corps.
la chaleur latente massique de fusion du corps.
Chaleur latente de vaporisation
-
Appareillage simple (appareil de Berthelot ; appareil de Richards)
Appareil de Richards |
Un vase Dewar est traversé par un tube
T ouvert à ses deux extrémités. Ce tube se raccorde
à un serpentin S plongé dans un calorimètre adiabatique.
Le serpentin aboutit à un réservoir de condensation R et
communique avec latmosphère par le tube A . Lébullition
du liquide placé dans le vase Dewar a lieu ainsi sous la pression
atmosphérique.
Des gouttes liquides peuvent provenir dune légère condensation
qui se produit dans la vapeur en montant dans le vase Dewar. Elles sont
vaporisées à nouveau lorsquelles traversent le fond dans
le tube T et la vapeur arrivant dans S est sèche.
Légalité de température entre le calorimètre
et lenceinte E est obtenue en faisant tomber de lacide dans la solution
de soude contenue dans E. |
 |
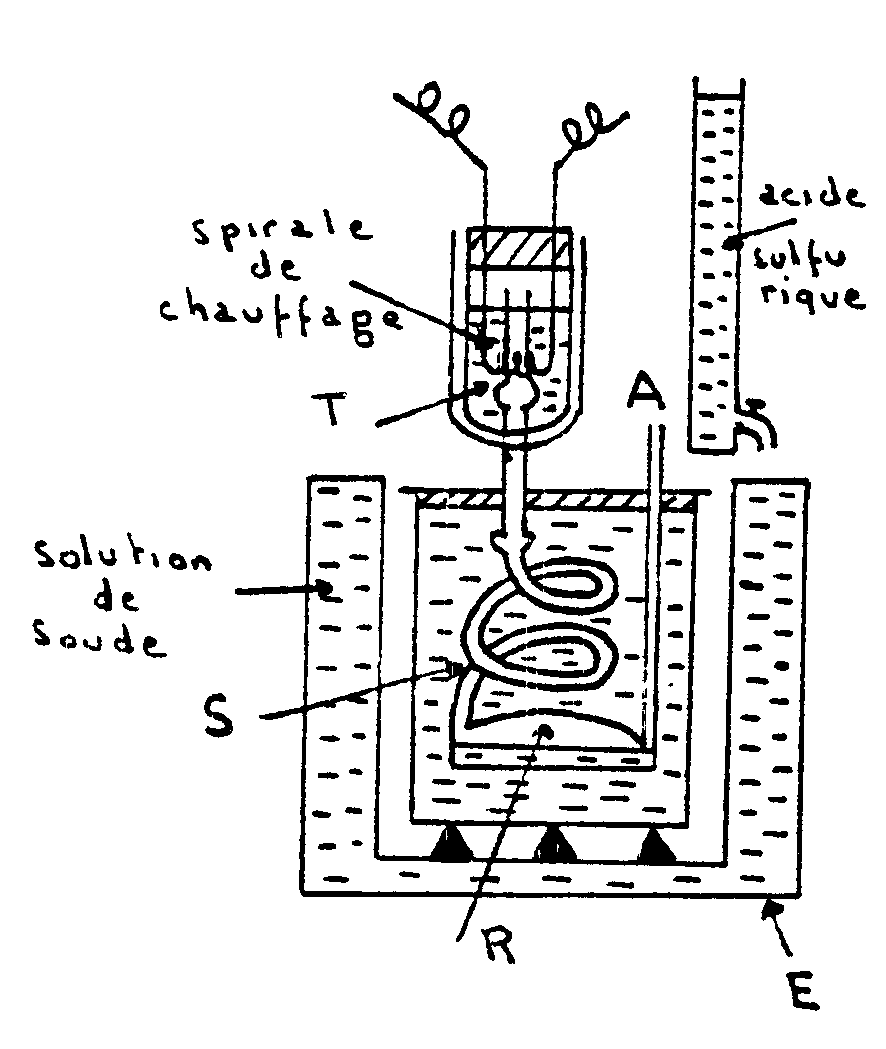
Lappareil est entièrement clos et la
pression intérieure est obtenue grâce à une atmosphère
artificielle (réservoir de 600 litres pouvant supporter des pressions
inférieures à 20 atmosphères). Un robinet R permet
de mettre la chaudière en communication soit avec un condenseur
C, soit avec un serpentin de condensation S.
Au début, la chaudière communique avec le condenseur
et on chauffe le liquide (eau) jusquà ébullition ; les vapeurs
viennent se condenser dans C. |
Quand la distillation est devenue régulière, on met la
chaudière en communication avec le serpentin. La vapeur sèche
qui arrive sy condense et quand lexpérience a duré assez
longtemps, on remet la chaudière en communication avec le condenseur,
puis on recueille avec le robinet r leau condensée en S.
Désignons par m la masse du liquide condensé, 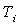 sa capacité calorifique massique,
sa capacité calorifique massique, 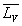 sa chaleur latente de vaporisation sous la pression considérée
à la température
sa chaleur latente de vaporisation sous la pression considérée
à la température 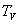 et la valeur
en eau du calorimètre dont la température initiale est
et la valeur
en eau du calorimètre dont la température initiale est 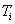 et la température finale
et la température finale 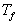 .
.
Le bilan énergétique sécrit : 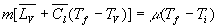
Cette méthode se prête très facilement à la
mesure des chaleurs de vaporisation.
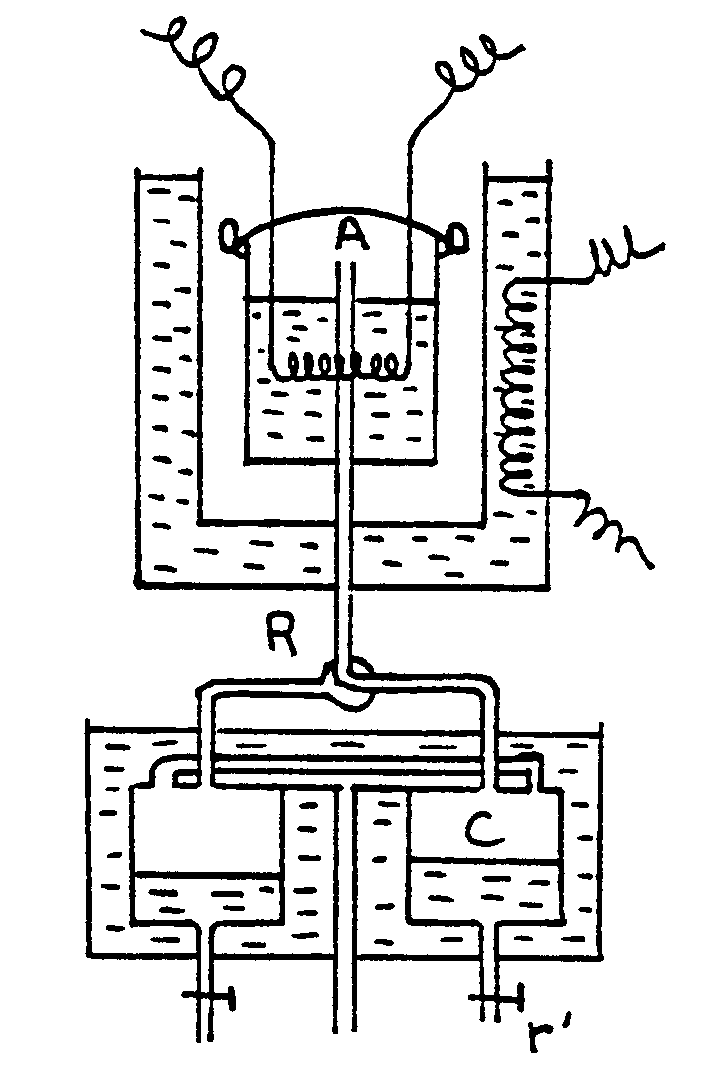 |
On produit la vaporisation par le passage dun
courant dintensité I dans une résistance R
immergée dans le liquide.
Si, pendant le temps  ,
on vaporise, en régime permanent à la température ,
on vaporise, en régime permanent à la température  ,
une masse de liquide m, on a léquation ,
une masse de liquide m, on a léquation 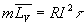 . .
La vaporisation est produite dans une enceinte close A, très
robuste. La vapeur se dégage par un ajustage muni dun robinet R.
Elle se condense dabord dans un condenseur auxiliaire jusquà ce
que lon obtienne un régime permanent. La vapeur est ensuite condensées
dans C, recueillie par le robinet r et pesée.
Soit m la masse du liquide vaporisé, m celle de la
vapeur qui est sortie de la chaudière (que lon a recueillie par
condensation),  les volumes massiques du liquide et de la vapeur.
les volumes massiques du liquide et de la vapeur. |
Laugmentation de volume 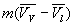 produite par la vaporisation est égale au volume de vapeur
produite par la vaporisation est égale au volume de vapeur 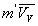 sorti
de la chaudière ð
sorti
de la chaudière ð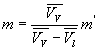 .
.
m diffère dautant plus de m que lécart  est plus faible cest à dire que lon sapproche de la température
critique.
est plus faible cest à dire que lon sapproche de la température
critique.
Le tableau ci-après donne les valeurs pour leau.
|
Températures
|
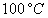
|
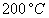
|
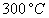
|
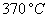
|
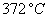
|
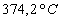
|
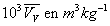
|
1,673
|
0,1272
|
0,02162
|
0,00500
|
0,00450
|
0,00323
|
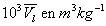
|
0,001043
|
0,001156
|
0,001404
|
0,00223
|
0,00238
|
0,00323
|
2.3. Résultats
Chaleur latente de fusion
Sous la pression atmosphérique, pour différents corps
:
- 333 kj/kg pour leau à 0 °C,
- 20,5 kj/kg pour le phosphore à 44 °C,
- 22,5 kj/kg pour le plomb à 327 °C.
Chaleur latente de vaporisation
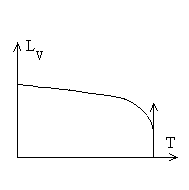 |
Les chaleurs latentes de vaporisation diminuent
avec la température et atteignent une valeur nulle au point critique
puisque phases liquide et gazeuse sont identiques.
On utilise souvent des lois empiriques du type 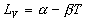 dans
un intervalle de température. dans
un intervalle de température.
Ainsi pour leau entre 100 et 200 °C,
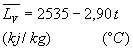 |
Nous donnons ci-après quelques chaleurs latentes massiques de
vaporisation exprimées en kj/kg sous une pression dun atmosphère.
Ammoniac 1425 ; Benzène 393 ; Dioxyde de Carbone 594 ; Eau 2253
; Dioxyde de Soufre 397 ; Ethanol 903
Limportances des valeurs des chaleurs latentes et, plus particulièrement,
de celles de vaporisation justifie lutilisation des changements de phase
dans beaucoup de machines thermiques.
Les chaleurs latentes de fusion, de sublimation et de vaporisation sont
positives, celles de solidification, de condensation à létat
solide et de condensation à létat liquide sont respectivement
opposées.