Description macroscopique de la matière
Plan
1. Physique des milieux continus
2. Statique des fluides
3. Température et équation d'état
des gaz parfaits
4. Equations d'état
5. Déformation des solides
6. Changements de phases (d'état) des corps
purs
1. Physique des milieux continus
Parce que lenseignement des notions datomistique est largement
développé, chacun est convaincu du caractère discontinu
de la matière. Pourtant à léchelle macroscopique
courante, la matière nous parait continue. Ces deux façons
dappréhender la matière sont complémentaires, se
nourrissent lune de lautre tout au long de lenseignement des Sciences
Physiques.
La Physique ne sest pas faite en un jour, elle est le résultat
de tâtonnements successifs, un cours nest jamais linéaire.
Il arrive souvent que lon soit amené à faire appel au sens
commun pour présenter une notion, un modèle qui, de fait,
nous sert à définir, à qualifier, à quantifier
le phénomène
donc à le comprendre
Macroscopiquement aussi petit soit-il, un échantillon de matière,
en Physique des milieux continus, contient un grand nombre de particules - 3
cm3 deau renferme 1023
molécules - . Lhypothèse de milieu continu considère que
la matière occupe continûment tout le volume. Ainsi si un échantillon
de volume dV contient des particules, la masse dm de léchantillon
sera la somme des masses des particules et, en Physique des milieux continus,
on définit la masse volumique ![]() qui a donc un caractère de valeur moyenne.
qui a donc un caractère de valeur moyenne.
1.1. Systèmes. Variables détat. Etat déquilibre. Equations détat.
Nous appelons système un ensemble constitué dun
grand nombre de particules. Tout système sera en interaction avec
dautres systèmes de lUnivers qui constituent le
milieu extérieur.
Linteraction se traduit par des échanges de matière
et des échanges dénergie sous forme de travail et de chaleur.
Système et milieu extérieur peuvent ne pas présenter
de différences de nature, mais lobservateur les différencie
par le rôle quil leur fait jouer, les frontières les séparant
pouvant être réelles ou fictives.
Un système est fermé sil ne peut échanger
de la matière avec lextérieur, dans le cas inverse il est
ouvert.
Un système fermé est isolé sil néchange
aucune énergie avec lextérieur, il nest pas isolé
sil échange de lénergie.
En Physique des milieux continus, on décrit létat dun
système par lintroduction dun nombre restreint de paramètres
mesurables qui rendent compte de létat du système. On
les appelle variables détat.
Cest lexpérience qui est déterminante pour définir
les variables détat nécessaires.
La température, la pression, le volume,
la quantité de matière sont les variables détat
les plus couramment nécessaires.
Un système est en état déquilibre si les
variables détat du système sont constantes (dans le temps)
et uniformes dans toute partie homogène du système.
Pour mieux comprendre : Soit le mur extérieur dune maison chauffée. Sa température varie dun endroit à un autre du mur, de la température intérieure à celle extérieure. Le système " mur " est en état de déséquilibre même si, en chaque endroit, la température est constante. Dans ce cas, on dira que le mur est en régime permanent. Si la température évoluait au cours du temps, le mur serait en régime transitoire.
Dans un système en état déquilibre, on
appelle
équation détat toute relation entre les variables
détat.
En Physique des milieux continus, la Thermodynamique est définie
comme la science qui étudie les phénomènes où
intervient la température. Elle est née vers les années
1820, au début de lère industrielle, de la nécessité
de connaître, sur les machines thermiques construites, la relation
entre les phénomènes thermiques et les phénomènes
dynamiques,
doù son nom.
1.2. Transformations dun système
Dans une transformation il y a variation dau moins une variable
détat du système qui évolue dun
état initial
à un état final. Sauf pour les systèmes fermés,
isolés où il y a transformation spontanée du système
vers un état déquilibre si celui-ci nétait pas en
état déquilibre, la transformation se produit par laction
du milieu extérieur.
Pour que la transformation puisse être définie convenablement,
il conviendra que les états initial et final soient des états
déquilibre.
Si état initial et état final sont identiques,
la transformation est appelée cycle.
Les différents types de transformations
Transformations quasistatiques
Si dans une transformation, le système reste, à chaque instant, très voisin dun état d équilibre, la transformation est dite quasistatique. A chaque instant, en première approximation, les variables détat restent définies.
Transformations réversibles
Une transformation est dite réversible si le système évolue
en passant par une suite continue détats déquilibre
et
sil existe une transformation permettant de ramener le système
et le milieu extérieur, à chaque instant, à léquilibre
précédent. De fait, cette transformation ramène système
et milieu extérieur dans leurs états initiaux.
Il y a contradiction entre état déquilibre et évolution,
en toute rigueur une transformation réversible nest pas réalisable.
On peut, au mieux, sen approcher par une transformation quasistatique
avec possibilité de revenir à létat précédent.
Transformations irréversibles
Ce sont toutes celles qui ne remplissent pas les conditions de la réversibilité.
Remarque
A chaque instant dune transformation réversible, voire quasistatique, on peut écrire les équations détat du système. Il existe, en plus, une (ou plusieurs) relation(s) entre les variables détat liées à la nature de la transformation. Il ne faut pas donner un statut déquation détat à cette (ces) dernière(s) relation(s).
2.1. Pression dans un fluide au repos (en équilibre dans un référentiel galiléen)
Notre sens commun nous fait appréhender un fluide (gaz ou liquide) comme étant de nature très différente dun solide. Ce dernier a une forme propre qui nous permet de le reconnaître. Liquide ou gaz nont pas de forme propre, ils épousent la forme du récipient qui les contient, sont déformables sous la moindre action. Liquide et gaz ont des différences : un liquide, contrairement à un gaz, a un volume défini, il ne remplit pas tout le volume du récipient.
2.1.1. Etude expérimentale dans le champ de pesanteur
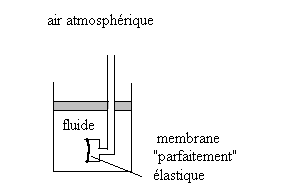 |
|
La forme de la membrane est indépendante de lorientation de
la membrane autour dun même point, les efforts sur la membrane sont
perpendiculaires à celle-ci.
Si le fluide est un liquide, la déformation de la membrane augmente
de manière significative avec la profondeur.
Autre expérience : si, dans un récipient contenant un liquide, nous perçons un trou, lécoulement de liquide se produit perpendiculairement à lorifice quelque soit lorientation de ce dernier.
2.1.2. Définition de la pression dans un fluide
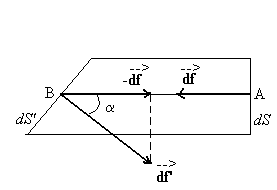 |
Considérons un fluide en équilibre
dans un référentiel lié au sol supposé galiléen
et une portion de ce fluide limitée extérieurement par un
cylindre élémentaire de révolution daxe AB horizontal
et dont les bases sont, en A une section droite dS, en B la surface plane
dS dont la normale fait un angle a avec laxe
AB.
Nous avons |
Cette portion de fluide nest pas soumise quà son seul poids
(voire à dautres forces dues à un champ extérieur)
pour être en équilibre. Elle est soumise à des forces
" superficielles " (sur les surfaces délimitant le cylindre) dues
au fluide extérieure environnant la portion, les forces internes
à la portion ninterviennent pas car elles obéissent au principe
de laction et de la réaction.
Les forces ![]() exercées
par le fluide environnant sur les bases (non matérielles) dS et dS du
volume élémentaire de fluide sont normales à celles-ci
(sinon il y aurait glissement entre les couches du fluide puisquun fluide se
déforme à la moindre action extérieure).
exercées
par le fluide environnant sur les bases (non matérielles) dS et dS du
volume élémentaire de fluide sont normales à celles-ci
(sinon il y aurait glissement entre les couches du fluide puisquun fluide se
déforme à la moindre action extérieure).
La condition déquilibre sur laxe horizontal se traduit par ![]() .
.
Soit ![]() qui,
par définition, est la pression dans le fluide.
qui,
par définition, est la pression dans le fluide.
La pression p en un point dun fluide en équilibre est indépendante
de lorientation de lélément de surface qui sert à
la définir.
La pression est une grandeur scalaire positive, la force de pression
est une grandeur vectorielle.
La pression est la même en tous les points dun plan horizontal
pris dans un fluide en équilibre.
2.1.3. Relation locale traduisant léquilibre dun fluide dans un référentiel galiléen
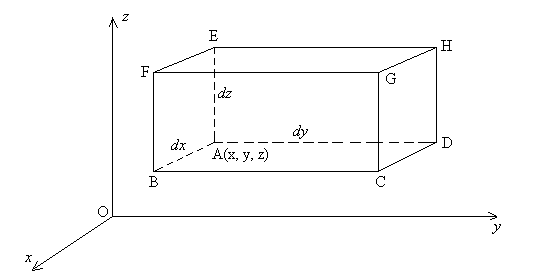
Exprimons la relation déquilibre pour un élément
de fluide en forme de parallèpipède rectangle de dimensions
dx,
dy et dz à partir du point A de coordonnées
x,
y et z.
Sur la surface rectangulaire ABFE, nous avons une pression ![]() et, sur la surface rectangulaire DCGH, nous avons une pression
et, sur la surface rectangulaire DCGH, nous avons une pression ![]() .
.
Il en est de même sur les autres faces du parallèpipède
rectangle en permutant les coordonnées. Les forces dues à des
champs de forces extérieures sécrivent ![]() .
.
En appliquant les relations déquilibre de la Mécanique et en
projetant suivant la direction Oy, on obtient ![]() .
.
Soit encore ![]() ;
; ![]() ;
; ![]()
Sous forme vectorielle, ces trois dernières relations sécrivent
:
![]()
Remarque : le gradient de pression nest due quaux champs de forces extérieures.
2.2. Cas dun fluide au repos dans le champ de pesanteur
Principe de lhydrostatique (dit de Pascal)
Le référentiel est un référentiel lié au
sol terrestre, le fluide a pour masse volumique m
et le champ de pesanteur (qui tient compte de l'accélération d'entraînement
due à la rotation de la Terre due autour de l'axe Sud-Nord) est le seul
champ de forces extérieures.
Dans ce cas ![]() relation équivalente à
relation équivalente à ![]() si laxe
si laxe ![]() est
vertical ascendant. Ceci constitue le principe de lhydrostatique ou
principe de Pascal.
est
vertical ascendant. Ceci constitue le principe de lhydrostatique ou
principe de Pascal.
Autre démonstration :
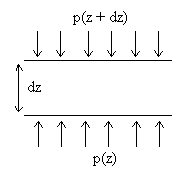 |
Dans le champ de pesanteur, on considère une
tranche de fluide à l'altitude z d'épaisseur L'axe des z est vertical ascendant, on appelle L'écriture du principe fondamentale de la dynamique conduit à |
Laugmentation de pression entre une altitude z + dz et une altitude
z
est due au poids du fluide dans le cylindre de hauteur verticale
dz
et de surface horizontale unitaire.
Lévolution de pression est donc continue, même à
la séparation de deux fluides.
La pression a une altitude z est égale au poids, par
unité de surface horizontale, des couches de fluide situées
à la verticale au-dessus de z.
Dans un même fluide au repos, les surfaces dégale altitude
sont isobares (même pression).
Dans le système MKSA, les pressions se mesurent en N m-2
que lon appelle Pascal (Pa).
Attention, il ne faudrait pas conclure que les
forces de pression sexercent verticalement. Elles sexercent perpendiculairement
à tout élément de surface.
2.2.1. Surface de séparation entre deux fluides non miscibles au repos
Considérons deux points de cette surface. Soit dz leur
différence daltitude.
On peut écrire, en négligeant toute discontinuité
de la pression à la traversée de la surface de séparation
:
![]() ce qui entraîne
dz = 0.
ce qui entraîne
dz = 0.
La surface de séparation entre deux fluides non miscibles
au repos est plane et horizontale.
Létude de la stabilité de léquilibre montrerait que le liquide de densité plus faible se place au dessus de celui de densité plus forte.
2.2.2. Fluide incompressible (liquide)
La masse volumique est constante et lintégration du principe de lhydrostatique
donne ![]()
Applications : baromètre - presse hydraulique

Dans un baromètre à mercure, pour une pression atmosphérique
normale, la hauteur h est égale à 760 mm ce qui correspond à ![]()
Souvent, les pressions sont exprimées en mm de mercure, en atmosphère
( 1 atm correspond à la pression atmosphérique normale),
en bar (1 bar = 105 Pa) ou millibar (mbar).
Nous remarquerons que pour une hauteur deau de 3m, la variation de
pression est égale à 30000 Pa soit à peu près
le tiers de la pression atmosphérique normale.
A léchelle humaine courante, les variations de pression
sont sensibles dans les liquides.
Un baromètre mesure une pression absolue puisque la pression dans le vide est nulle (pas de masse = pas de poids). Les manomètres mesurent les différences de pression.
2.2.3. Fluide compressible (gaz)
La masse volumique dépend de la pression et nous le verrons de
la température. On ne peut intégrer directement la relation
dp
= -mg dz.
Cependant les masses volumiques des gaz sont faibles (air dans les
conditions courantes 1,3 Kg m-3)
et, à léchelle humaine courante, on négligera
les variations de pression avec laltitude dans les gaz.
Seul lair atmosphérique présente des différences
daltitude suffisantes pour ne pas négliger les variations de pression.
2.2.4. Théorème dArchimède et corps flottants
Théorème dArchimède
Considérons (Fig. a) un corps entièrement immergé dans un fluide homogène au repos. Il occupe un volume V et subit de la part du fluide des forces de pression.

" Tout corps plongé dans un fluide en équilibre est soumis
de la part de celui-ci à une poussée verticale, dirigée
de bas en haut, égale au poids du liquide de remplacement et appliquée
à son centre de masse appelé centre de carène. "
En effet, en labsence de corps immergé, le fluide de remplacement
serait en équilibre sous laction des forces de pression exercées
par le fluide environnant et des forces de pesanteur. Les forces de pression
sont lopposé du poids du fluide de remplacement.
Autre démonstration : nous appliquons le théorème
du gradient (cas particulier du théorème dOstrogradsky)
pour la résultante des forces de pression sur une surface fermée
S
contenant le volume V.
![]() (le vecteur
(le vecteur ![]() est orienté vers lextérieur de la surface fermée).
est orienté vers lextérieur de la surface fermée).
Ce théorème reste vrai si le corps est immergé dans plusieurs fluides en équilibre.
Statique des corps flottants
Lorsque le poids du liquide de remplacement est supérieur au
poids du corps, seule une partie de ce dernier est immergée. Le
corps est alors soumis à deux forces : son poids, appliqué
au centre de masse C, et la poussée dArchimède appliquée
au centre de carène K.
A titre dexemple, considérons léquilibre dune
boîte parallélépipédique à section rectangulaire
ouverte flottant sur leau (Fig. b). Une telle boîte, de dimensions
L
= 10 m, l = 4 m, h = 3 m et de masse M = 20
t,
constitue un modèle simplifié dune embarcation flottant
sur leau. Elle senfonce dans leau dune hauteur h telle que
:
![]()
Notons que, la boîte étant ouverte et les parois latérales
de masse négligeable, le centre de masse C est situé au centre
de la base rectangulaire, alors que le centre de carène K est au-
dessus de lui à la distance h/2 . Dans ce cas, léquilibre
est stable : une légère rotation de la boîte autour
de laxe longitudinal passant par le centre de masse produit des oscillations
autour de la position déquilibre.
Létude de la stabilité de la position déquilibre, évidemment capitale dans la construction des navires, nest pas envisagée.
2.3. Relation locale traduisant léquilibre dun fluide dans un référentiel non galiléen
En Mécanique, on apprend que le mouvement dune masse " ponctuelle "
m (mieux : le mouvement du centre de masse dun système
matériel de masse totale m) est régi, dans un référentiel
non galiléen, par la relation ![]() où on introduit les forces dentraînement et de Coriolis.
où on introduit les forces dentraînement et de Coriolis.
Labsence de mouvement relatif imposent accélération et vitesse
relatives nulles et donc ![]() et
et ![]()
Par suite, la relation déquilibre sécrit ![]()
Pour le champ de forces extérieures de pesanteur ![]()
2.4. Le phénomène de tension superficielle
Une molécule au sein dun liquide est soumise de la part des autres
molécules à des forces de Van der Waals dorigine électromagnétique
qui varient en![]() . Ce
type dinteraction décroît très rapidement et peut être
négligé au-delà dune distance de lordre dune dizaine
de nanomètres.
. Ce
type dinteraction décroît très rapidement et peut être
négligé au-delà dune distance de lordre dune dizaine
de nanomètres.
A la surface de séparation de deux fluides (liquide-gaz ou liquide-liquide),
la situation est différente en particulier lisotropie des forces
qui existe au sein dun fluide nest plus la règle et il apparaît
un système de forces différent dans la couche capillaire
(couche de liquide proche de la surface de séparation de deux fluides
et dont lépaisseur est de lordre du nanomètre).
A titre dexemples, chacun visualisera que lon peut soulever une pellicule
deau savonneuse avec un anneau et quen soufflant sur cette pellicule
on peut former un bulle de savon dont la surface en forme de sphère
est tendue.
2.4.1. Tension superficielle
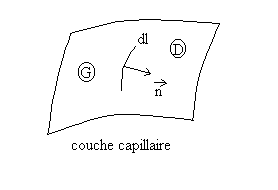 |
Les phénomènes observés
suggère de traiter la couche capillaire comme une membrane élastique
supposée dépaisseur nulle.
Effectuons une incision de longueur dl dans la couche capillaire. Les lèvres sécartent ce qui signifie que, préalablement à lincision, la partie droite D de la couche capillaire exerce sur lélément dl appartenant à la partie de gauche G une force attractive |
|||
|
|
|
|
||
| eau |
|
|
||
| éthanol |
|
|
Remarque : la partie droite D de la couche capillaire peut être remplacée par un solide ; la force garde la même valeur | |
| glycérine |
|
|
||
| eau |
|
|
||
2.4.2. Loi de Laplace
| Plongeons un tube de faible diamètre
(capillaire) dans un liquide contenue dans une cuve de grandes dimensions.
Le liquide sélève dans le tube. Ce phénomène sappelle capillarité et la loi de Jurin permet den rendre compte. A partir de loi de Laplace, on montre la loi de Jurin : |
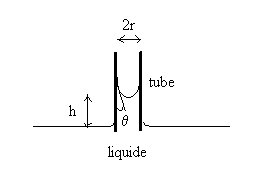 |
Langle q de contact dépend de linterface : il est de 0° pour les interfaces eau-verre propre et éthanol-verre propre et respectivement de 140°, 90° et 107° pour les interfaces mercure-verre, eau-argent et eau-paraffine.
2.4.4. Mesure des tensions superficielles
Il existe plusieurs méthodes de mesure des tensions superficielles. Nous citerons celle utilisant directement la loi de Jurin, celle utilisant larrachement dun solide du liquide et celle utilisant le détachement des gouttes de liquide dun tube.
3. Température et équation détat des gaz parfaits
" Il fait chaud ", " il fait froid ", " cest chaud ", " cest froid
" ... que dexpressions du langage courant pour traduire le fait que la
température fait partie du quotidien humain.
La première notion de température est physiologique,
sensitive. Cependant, nos sensations sont insuffisantes pour établir
une échelle de température et comparer des températures.
Lobservation, lexpérience nous apprennent quun même
système change (par exemple changement de volume) lorsque nos sensations
de température évoluent. Ce constat va nous permettre de
préciser quantitativement le paramètre physique température.
3.1. Principe de Thermométrie (ou Principe 0 de la Thermodynamique)
Considérons deux corps isolés en état déquilibre. Mis en contact thermique entre eux (à ce stade de nos connaissances en contact physique) ils évoluent vers de nouveaux états déquilibre.
Le principe 0 de la Thermodynamique affirme que la variable détat
température est caractéristique des états déquilibre
atteints.
En dautres termes et en raisonnant de proche en proche, des systèmes
mis en contact thermique évoluent vers des états déquilibre
où ils ont même température.
3.2. Echelles de température. Echelle légale
3.2.1. Thermométrie et grandeur thermométrique
La température nest associé à aucune loi physique,
nous pouvons donc choisir des valeurs arbitraires correspondant à
des
états déquilibre déterminés. Ainsi la température
peut être la valeur dune grandeur physique (ou une fonction monotone
arbitraire de cette grandeur) dun système à condition détablir
une correspondance bi-univoque entre grandeur physique et température.
Par comparaison des états déquilibre dautres systèmes
avec ceux du système choisi, on mesurera la température des
autres systèmes.
Le système choisi est appelé thermomètre,
la grandeur physique grandeur thermométrique.
Exemple : le thermomètre est un fil, la grandeur thermométrique
sa longueur.
La longueur dépend de la température puisque, si je "
chauffe " le fil (je change ma sensation de toucher, je conclus que la
température a varié) je constate que la longueur varie.
La température peut être définie soit comme la
valeur de la longueur elle-même, soit comme une fonction monotone
arbitraire de cette longueur.
Il suit que pour rendre la notion de température quantitativement
utilisable par chacun, il a fallu définir une échelle
universelle cest à dire un thermomètre de référence,
une grandeur thermométrique et une fonction monotone entre cette
grandeur et la température.
La Science ne sest pas faite en un jour et la définition actuelle
de léchelle légale de température est le résultat
de tâtonnements successifs. Cest pourquoi, de nos jours, au quotidien,
coexistent léchelle Kelvin, léchelle Celsius, léchelle
Farenheit, seule léchelle Réaumur est tombée en désuétude.
3.2.2. Faits expérimentaux
Points fixes
Certains états déquilibre sont particulièrement
utiles car facilement reproductibles. On les appelle points fixes.
Il sagit des états déquilibre de deux phases dun même
corps pur sous une pression donnée (solide en fusion, liquide en
ébullition, ...) ou de trois phases dun même corps pur ce
qui se produit à une certaine pression (point triple).
Ces états déquilibre sont facilement reproductibles
car indépendants des masses des phases en présence.
Exemple : la résistance ou la longueur dun fil de platine
immergé dans un mélange de glace et deau sous une pression
donnée est indépendante des masses deau et de glace en présence
et reprend toujours la même valeur si on refait le mélange,
la pression étant inchangée.
Nous savons reproduire facilement des températures.
Le gaz parfait, limite de comportement de tous les gaz réels.
Lexpérience montre que, si lon trace les courbes de compressibilité isotherme pour un nombre donné de molécules, on obtient dans un diagramme p, pV (dit diagramme dAmagat) les courbes ci-après.
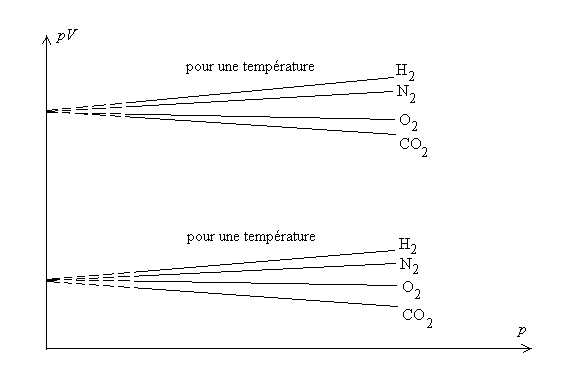
![]() a la même valeur
pour une température q fixée.
Cette limite est proportionnelle au nombre de molécules N ou de
moles n ( N = nN
où N = 6,023 1023
est le nombre dAvogadro). Cette limite est indépendante du gaz considéré.
a la même valeur
pour une température q fixée.
Cette limite est proportionnelle au nombre de molécules N ou de
moles n ( N = nN
où N = 6,023 1023
est le nombre dAvogadro). Cette limite est indépendante du gaz considéré.
Ainsi ![]() pour tous
les gaz.
pour tous
les gaz.
Définition du gaz parfait : Un gaz qui aurait,
quelle que soit la pression, le comportement de tout gaz réel à
pression nulle est appelée gaz parfait.
Les variables détat pression p, volume V, température
q et nombre de moles n sont liées
par léquation détat ![]()
3.2.3. Echelle légale de température
Le Kelvin : proposé en 1954, adopté en 1967 par
le Comité International des Poids et Mesures, le Kelvin est défini
de la manière suivante :
- Le thermomètre de référence est le thermomètre
à gaz parfait,
- La grandeur thermométrique est le produit pV,
- La fonction monotone est linéaire soit pV = nRT où
le symbole T est réservé à la température
Kelvin (appelée aussi absolue) et où R est la constante
des gaz parfaits.
La valeur de R est déterminée à partir dun
point fixe de référence (celui de léquilibre des
trois phases solide, liquide et gazeuse du corps pur H2O
appelé point triple de leau). Pour ce point fixe, on a donné
la valeur 273,16 à la température. La mesure de pV
à cette température, pour une mole, par extrapolation jusquà
la pression nulle des courbes de compressibilité isotherme des gaz
réels donne 2271,1 joules.
Il suit que R = 2271,1/273,16 = 8,314 MKSA.
La notation symbolique du Kelvin est K.
Remarque : pV = nRT équation liant
les variables d'état des gaz parfaits est appelée équation
d'état des gaz parfaits. Historiquement, cette relation est
connue sous le nom de loi de Boyle-Mariotte.
Bien que valable en toute rigueur à la pression nulle, cette
équation sapplique avec des précisions très satisfaisantes
jusquà des pressions de quelques atmosphères pour les gaz
réels.
3.2.4. Autres échelles de température
Pour les échelles Celsius (notation t), Farenheit (notation
F)
et Réaumur (notation R), seule diffère dans la définition
la fonction monotone qui reste linéaire mais de la forme pV =
aq + b.
Les correspondances sont souvent établies de la manière
suivante :
![]()
où ![]() correspondent respectivement aux points fixes débullition de leau et
de fusion de la glace sous la pression atmosphérique normale, les températures
respectives étant 373,15 K et 273,15 K soient encore 100
°C et 0°C.
correspondent respectivement aux points fixes débullition de leau et
de fusion de la glace sous la pression atmosphérique normale, les températures
respectives étant 373,15 K et 273,15 K soient encore 100
°C et 0°C.
Il y a une simple translation entre les échelles Celsius et
Kelvin (t = T - 273,15).
3.3. La mesure des températures
Mis à part quelques laboratoires spécialement équipés,
on ne mesure pas une température avec un " thermomètre à
gaz parfait ".
On se sert de thermomètres (et grandeurs thermométriques)
que lon étalonne, dans la gamme de température dutilisation,
à partir des valeurs des températures de points fixes déterminées
par ces quelques laboratoires.
3.3.1. Points fixes fondamentaux
On trouvera ci-après les températures officielles assignées
à certains points fixes ; ces valeurs, sauf pour les points triples,
correspondent à des états déquilibre sous la pression
atmosphérique normale.
|
|
|
|
| Point d'ébullition de l'hélium |
|
|
| Point triple de l'hydrogène |
|
|
| Point d'ébullition de l'hydrogène à 33330,6 Pa |
|
|
| Point d'ébullition de l'hydrogène |
|
|
| Point d'ébullition du néon |
|
|
| Point triple de l'oxygène |
|
|
| Point d'ébullition de l'oxygène |
|
|
| Point de fusion de l'eau |
|
|
| Point triple de l'eau |
|
|
| Point d'ébullition de l'eau |
|
|
| Point de fusion du Zinc |
|
|
| Point de fusion de l'argent |
|
|
| Point de fusion de l'or |
|
|
3.3.2. Les thermomètres
 |
Trois fils constitués de deux métaux
ou alliages différents M et M sont soudés (ou en contact)
en a et b où règnent des températures Des relations de type Leur domaine dutilisation varie entre -180 et 2500 °C. |
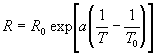
3.3.3. Thermométrie
Mesurer une température savère souvent difficile ; en
effet, faire un étalonnage précis dun thermomètre
soulève des problèmes, introduire le thermomètre à
lendroit où lon veut connaître la température perturbe,
parfois notablement, la température.
Nous nen dirons pas plus dans le cadre de ce cours.
4.1. Applications simples de léquation détat des gaz parfaits
4.1.1. Un gaz parfait obéit aux lois de Gay-Lussac et de Charles
On dit quun gaz obéit à la loi de Gay-Lussac si, à
pression constante, son volume est proportionnel à la température.
On dit quun gaz obéit à la loi de Charles si, à
volume constant, sa pression est proportionnelle à la température.
Pour un gaz parfait à ![]() (Loi de Gay-Lussac)
(Loi de Gay-Lussac)
Pour un gaz parfait à ![]() (Loi de Charles)
(Loi de Charles)
4.1.2. Un gaz qui obéit aux lois de Gay-Lussac et de Charles est un gaz parfait
Loi de Gay-Lussac : à ![]() .
Si on change la valeur de p, cette dernière constante change.
Cest donc une fonction de
.
Si on change la valeur de p, cette dernière constante change.
Cest donc une fonction de ![]()
Loi de Charles : à ![]() .
Si on change la valeur de V, cette dernière constante change.
Cest donc une fonction de
.
Si on change la valeur de V, cette dernière constante change.
Cest donc une fonction de ![]()
En réunissant ces deux résultats, on obtient ![]() .
.
Soit ![]() puisque
la relation doit être vraie quels que soient V et p.
puisque
la relation doit être vraie quels que soient V et p.
![]() soit encore pV
= CT
soit encore pV
= CT
4.1.3. Mélange idéal de gaz parfaits
Dans une enceinte de volume V, nous mélangeons différentes
substances chimiques en phases gazeuses sans possibilité de réactions
chimiques entre elles. Le mélange est dit idéal.
Si p la pression du mélange est suffisamment faible,
lexpérience montre que :
![]() est le nombre
de moles du gaz i et n le nombre total de moles gazeuses sans distinction
de la substance gazeuse.
est le nombre
de moles du gaz i et n le nombre total de moles gazeuses sans distinction
de la substance gazeuse.
Le mélange se comporte comme un gaz parfait.
Si le gaz i était seul dans lenceinte, il se comporterait comme un
gaz parfait et sa pression ![]() serait donnée par la relation
serait donnée par la relation ![]() .
.
On remarquera que la pression p dans le mélange est liée
aux pressions ![]() par la relation
par la relation ![]() .
.
![]() est appelée
pression partielle du gaz i dans le mélange. La pression du mélange
est la somme des pressions partielles.
est appelée
pression partielle du gaz i dans le mélange. La pression du mélange
est la somme des pressions partielles.
On appelle fraction molaire du gaz i dans le mélange, la quantité ![]() .
.
On déduit que ![]() .
.
La quantité ![]() représente la masse totale du mélange si
représente la masse totale du mélange si ![]() est la masse molaire du gaz i.
est la masse molaire du gaz i.
On définit la " masse molaire du mélange " par 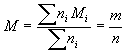 .
.
4.1.4. Autre écriture de léquation détat des gaz parfaits
pV = nRT sécrit aussi pV = (m/M) RT = mrT où r = R/M.
Si N est le nombre de molécules du gaz, N = nN
où ![]() N
est le nombre dAvogadro.
N
est le nombre dAvogadro.
PV = nRT![]() pV =
NkT où
pV =
NkT où ![]() est
la constante de Boltzmann.
est
la constante de Boltzmann.
Léquation détat des gaz parfaits sécrit encore ![]() en introduisant
en introduisant ![]() la densité volumique molaire et
la densité volumique molaire et ![]() la densité volumique des molécules.
la densité volumique des molécules.
Remarque : localement la pression est proportionnelle à la densité des molécules
4.1.5. Masse volumique et densité des gaz parfaits
Soit m = m/V la masse volumique. On obtient ![]() (autre
écriture de léquation détat des gaz parfaits).
(autre
écriture de léquation détat des gaz parfaits).
On appelle densité dun gaz le rapport de la masse dun certain
volume de gaz sur la masse du même volume dair pris dans les mêmes
conditions de pression et de température.
![]() (en grammes)
(en grammes)
4.2. Equations détat des corps réels
4.2.1. Cas des gaz
 |
Les premières expériences de compressibilité
isotherme des gaz, dans des domaines de pression très limités
ne dépassant pas quelques atmosphères, ont été
faites au 17ème siècle par
Mariotte en France et par Boyle en Angleterre.
Plusieurs expérimentateurs (Regnault, Andrews, Cailletet et Amagat) ont repris ces études au 19ème siècle dans des domaines de pression de plus en plus élevés. Kamerlingh Onnes à Leyde a étendu les mesures au domaine des basses températures. |
Ces mesures ont été reprises au 20ème
siècle avec une précision accrue sur tous les gaz connus
dans des domaines de température extrêmes et pour des pressions
atteignant 1000 atmosphères.
De longues séries de mesures systématiques ont été
faites dans deux laboratoires spécialisés : en Europe celui
de Van Der Waals à Amsterdam sous la direction de Michels et aux
Etats -Unis au laboratoire du Massachusetts Institute of Technology sous
la direction de Beattie.
Nous présentons, à titre dexemple, une série
de résultats en coordonnées dAmagat p, pV pour le
diazote.
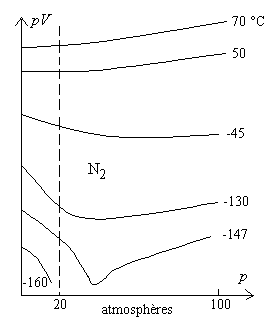 |
Au-dessous dune température (-147 °C
pour le diazote) les courbes sont interrompues car le gaz se liquéfie.
Cette température est appelée température critique
et notée TC .
Une autre température particulière apparaît, la température de Mariotte TM , où les courbes sont horizontales pour p = 0 ( Pour des températures supérieures à la température de Mariotte, les pentes pour p = 0 sont positives, elles sont négatives dans le cas contraire. Pour le gaz parfait, limite de comportement de tous les gaz réels lorsque la pression devient nulle, les courbes sont horizontales quelle que soit la température. Un gaz réel se rapproche au mieux du comportement du gaz parfait à sa température de Mariotte. |
Equations détat des gaz
Il nexiste pas déquation universelle rendant compte des courbes ci-dessus.
Pour des valeurs faibles de p, nous devons retrouver léquation
détat des gaz parfaits pV=nRT
Des équations classiques sont donc :
![]()
![]()
La validité de ces équations dépend, pour la précision
voulue, essentiellement du domaine de variation de la pression.
Ainsi jusquà 2 atmosphères, et même 10 ou 20, on utilise
léquation détat des gaz parfaits.
Léquation détat![]() est dune utilisation assez générale. Dans le chapitre " Description
microscopique de la matière ", on donnera une interprétation des
termes
est dune utilisation assez générale. Dans le chapitre " Description
microscopique de la matière ", on donnera une interprétation des
termes ![]() .
.
La plus célèbre des équations de ce type est celle de Van
der Waals :![]() où
où![]() (résultats expérimentaux).
(résultats expérimentaux).
Cette équation de Van der Waals interprète la température
de Mariotte.
![]()
Pour![]() , léquation
de Van der Waals sapproche au mieux de léquation détat des
gaz parfaits.
, léquation
de Van der Waals sapproche au mieux de léquation détat des
gaz parfaits.
Elle interprète aussi la température critique.
On utilise dautres équations détat de type Van
der Waals, par exemple
Diétérici ![]() ; Berthelot
; Berthelot ![]() ;
;
Clausius ![]()
Les équations de Van der Waals, Diétérici, Berthelot ou Clausius peuvent, dans certaines limites, sappliquer aux liquides.
4.2.2. Cas des liquides et des solides
En première approximation, le volume dun liquide ou dun solide
est indépendant de la température ou de la pression.
Cette approximation est souvent trop simplificatrice et il convient
de connaître léquation détat.
Les livres de données thermodynamiques présentent, dans
le cas des liquides et des solides, les coefficients thermoélastiques,
![]() coefficient de
dilatation à pression constante
coefficient de
dilatation à pression constante
![]() coefficient daugmentation
de pression à volume constant
coefficient daugmentation
de pression à volume constant
![]() coefficient de
compressibilité isotherme
coefficient de
compressibilité isotherme
Remarque : la relation mathématique ![]()
Le lecteur remarquera que lintégration de deux des coefficients thermoélastiques
fournit léquation détat et, inversement, la dérivation
de léquation détat donne les coefficients thermoélastiques
(pour un gaz parfait, ![]() ).
).
Nous ne décrirons pas les appareillages utilisables mais invitons
le lecteur intéressé à se reporter à des ouvrages
spécialisés.
Pour ce qui est de la connaissance du coefficient de dilatation à pression
constante des solides, on prend des solides en forme de fil et on mesure le
coefficient de dilatation linéaire à pression constante ![]() .
.
Le coefficient de compressibilité isotherme sobtient à
partir des modules de Young, de Poisson et/ou de cisaillement et cette
question sera abordé dans le paragraphe suivant " Déformation
des solides ".
Pour ce qui est de la connaissance du coefficient à pression
constante des liquides, la mesure se fait directement à partir de
lexpérience mise au point par Dulong et Petit ou à laide
dun dilatomètre à tige.
La mesure du coefficient de compressibilité isotherme pour les
liquides se fait directement dans des piézomètres à
capillaire.
Remarque : on ne mesure pas le coefficient b daugmentation de pression à volume constant parce quil est très difficile de faire subir à un liquide ou un solide une transformation isochore.
Equations détat des solides et des liquides
Les coefficients a etcTsont
faibles si bien que ![]() est
une équation détat habituelle dans des limites de température
et de pression à préciser pour chaque corps.
est
une équation détat habituelle dans des limites de température
et de pression à préciser pour chaque corps.
Un solide nest jamais parfaitement rigide : soumis à des forces
extérieures, il se déforme.
On distingue divers types de déformations : variation des dimensions,
flexion, cisaillement et torsion.
Il peut se faire quune partie de la déformation subsiste lorsque
laction extérieure cesse : on dit que le corps a subi une déformation
permanente
ou plastique.
Lorsque les forces extérieurs sont faibles et pour de nombreux
solides, la déformation disparaît lorsque laction extérieure
cesse : on dit que la déformation est élastique. Pour
chaque sollicitation et chaque corps, il existe une force limite au delà
de laquelle les déformations cessent dêtre élastiques,
cest la limite délasticité.
Nous limitons nos propos aux déformations élastiques
et présentons, dans le cas simple de solides homogènes et
isotropes, les relations entre les efforts intérieurs de cohésion
(contraintes) qui équilibrent les forces extérieures
et les déformations.
Allongement dun barreau
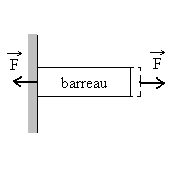 |
A ce solide dont lune des dimensions est grande
par rapport aux deux autres, appliquons sur les faces terminales, supposées
normales à la plus grande dimension, deux forces de traction de même
intensité F égales et opposées. Lexpérience montre que le barreau sallonge suivant la grande dimension et se contracte suivant les dimensions transversales. Ces variations de dimensions suivent une loi linéaire et réversible tant que |
La loi de Hooke permet décrire : ![]() où
où ![]() est
lallongement relatif du barreau, S la surface transversale,
est
lallongement relatif du barreau, S la surface transversale, ![]() la contrainte normale et
la contrainte normale et ![]() le module dYoung, coefficient caractéristique du matériau.
le module dYoung, coefficient caractéristique du matériau.
![]() a les dimensions
dune pression ; pour les métaux, il varie de
a les dimensions
dune pression ; pour les métaux, il varie de ![]() .
.
Corrélativement à cet allongement, se produit une contraction
des dimensions latérales (perpendiculaires à ![]() ).
).
Si l est une dimension latérale caractéristique, alors ![]() où
où ![]() est
le coefficient de Poisson. Ce coefficient est toujours inférieur
à 0,5 (de lordre de 0,3 pour les métaux, voisin de 0,5 pour le
caoutchouc).
est
le coefficient de Poisson. Ce coefficient est toujours inférieur
à 0,5 (de lordre de 0,3 pour les métaux, voisin de 0,5 pour le
caoutchouc).
Remarque : lexpérience a été décrite en
traction ; elle aurait pu être faite en compression, les résultats
étant symétriques dans la phase élastique.
5.2. Compression hydrostatique
On soumet un solide à une pression uniforme en limmergeant dans un
fluide. Une variation ![]() de la pression saccompagne dune variation
de la pression saccompagne dune variation ![]() du volume telle que
du volume telle que ![]()
Pour déterminer la relation entre![]() ,
nous prenons un solide en forme de parallélépipède rectangle
de cotés a, b et c.
,
nous prenons un solide en forme de parallélépipède rectangle
de cotés a, b et c.
La variation de pression ![]() sur la section (
sur la section (![]() )
provoque les déformations
)
provoque les déformations ![]() ,
, ![]() et
et ![]() .
.
On obtient des déformations de même forme pour les variations
de pression ![]() sur les sections (a, c) et (a, b).
sur les sections (a, c) et (a, b).
En superposant les déformations, on obtient au total :
![]()
ð![]() ð
ð ![]()
5.3. Contraintes tangentielles
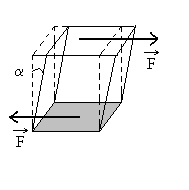 |
Sur les faces opposées dun parallélépipède
rectangle, on exerce des forces tangentielles dintensité F
égales et opposées. Le parallélépipède se déforme dun angle Tant que lon reste en deçà de la limite élastique, on peut écrire la loi |
G sexprime en ![]() et varie de
et varie de ![]() à
à ![]() pour les métaux.
pour les métaux.
Module dYoung, coefficient de Poisson et module de cisaillement
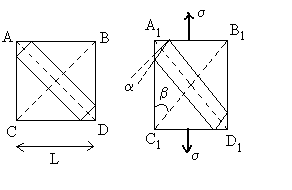 |
On considère un cube (trace carrée ABCD)
et on exerce sur ses faces supérieure et inférieure des contraintes
normales. Le cube se déforme et devient un parallélépipède
rectangle (trace rectangulaire Le parallélépipède rectangle, dépaisseur faible (trace rectangulaire de directions parallèles aux diagonales) se déforme sous laction des contraintes. |
Cette déformation peut être comprise comme due à des contraintes
tangentielles (cisaillement) dans les plans de direction ![]() .
.
Ces contraintes tangentielles sont égales à la projection
des contraintes normales, soit
![]()
Des considérations géométriques permettent décrire
que ![]() où
où ![]() représente la déformation du parallélépipède
rectangle.
représente la déformation du parallélépipède
rectangle.
 ð
ð![]() ð
ð![]()
5.4. Torsion
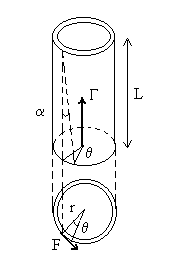 |
On considère un tube dépaisseur
faible e, de longueur L, de rayon r.
On lui fait subir une torsion dangle On se propose de calculer le moment La figure ci-contre montre une déformation de cisaillement dangle La force dF sur un élément de longueur dl
est égale à : |
En intégrant à tous les éléments de la base du
tube, on obtient ![]()
Pour un barreau cylindrique de même longueur, de rayon R,
on intègre la relation précédente en considérant
une succession de tubes dépaisseur ![]() .
.
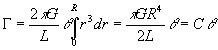
Remarque : évidemment, tube ou barreau exerce un couple résistant
(de rappel) opposé à celui que doit exercer lopérateur
extérieur pour effectuer la torsion.
5.5. Flexion
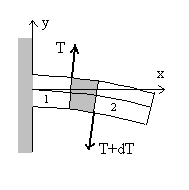 |
On considère une poutre (pour le dessin,
encastrée à lune de ses extrémités et libre
à lautre).
La poutre fléchit (sous leffet de son poids et déventuelles charges) et les génératrices, initialement suivant laxe des x deviennent des courbes déquation y(x). Il est clair que les génératrices supérieures sallongent, qque les génératrices inférieures se raccourcissent et quil existe une génératrice (fibre neutre) gardant la même longueur. |
Une tranche de poutre comprise entre labscisse ![]() subit de la part de la partie amont notée 1 et de la partie aval notée
2 des forces tangentielles (efforts tranchants), respectivement
subit de la part de la partie amont notée 1 et de la partie aval notée
2 des forces tangentielles (efforts tranchants), respectivement ![]() verticale ascendante et
verticale ascendante et ![]() verticale descendante, dont les effets sont de lempêcher de " tomber
" et de provoquer sa flexion (rotation).
verticale descendante, dont les effets sont de lempêcher de " tomber
" et de provoquer sa flexion (rotation).
![]() où
où ![]() représente les forces linéaires (par exemple poids) auxquelles
la poutre est soumise, soit léquation
représente les forces linéaires (par exemple poids) auxquelles
la poutre est soumise, soit léquation ![]() .
.
Ces forces tangentielles exercent un moment ![]() qui
est équilibré par les contraintes normales dues à lallongement
et au raccourcissement des génératrices situées de part
et dautre de la fibre neutre.
qui
est équilibré par les contraintes normales dues à lallongement
et au raccourcissement des génératrices situées de part
et dautre de la fibre neutre.
 |
|
Lensemble des forces ![]() est de direction pratiquement horizontale, leur sens est différent suivant
que lon considère une fibre située au dessus ou au dessous de
la fibre neutre.
est de direction pratiquement horizontale, leur sens est différent suivant
que lon considère une fibre située au dessus ou au dessous de
la fibre neutre.
Il ny a pas de déplacement suivant la direction de laxe des x,
aussi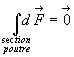 ð
ð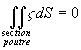 .
.
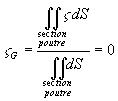 représente
le centre de gravité de chaque section de la poutre, ainsi la fibre
neutre est le lieu des centres de gravité de chaque section de la poutre.
représente
le centre de gravité de chaque section de la poutre, ainsi la fibre
neutre est le lieu des centres de gravité de chaque section de la poutre.
Lensemble des forces ![]() sur une
section de la poutre a un moment fléchissant
sur une
section de la poutre a un moment fléchissant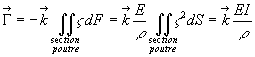 où
où 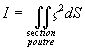 est
appelé moment quadratique.
est
appelé moment quadratique.
Léquation de la fibre neutre y(x) sobtient
en utilisant la formule du rayon de courbure :
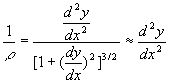 ð
ð![]() .
.
Les efforts tranchants se calculent à partir de ![]() ,
soit
,
soit ![]() ou de
ou de ![]()
Les constantes dintégration sont déterminées par
le système de fixation de la poutre,
- pour un encastrement horizontal en ![]() ,
on a
,
on a ![]() et
et ![]() [ cette dernière condition est imposée par le fait quun angle
non nul traduirait une cassure de la poutre,
[ cette dernière condition est imposée par le fait quun angle
non nul traduirait une cassure de la poutre,
- pour une extrémité libre en ![]() ,
on a
,
on a ![]() et
et ![]() ,
soient
,
soient ![]() et
et ![]() ,
,
- pour une fixation sur un point dappui en ![]() ,
on a
,
on a ![]() et
et ![]() puisque le point dappui empêche, là où il se trouve, le
déplacement et la flexion.
puisque le point dappui empêche, là où il se trouve, le
déplacement et la flexion.
La flèche de la poutre est la valeur maximale de y(x),
elle sera dautant plus faible que le matériau aura un module dYoung
de forte valeur et une forme telle que  soit le plus grand possible.
soit le plus grand possible.
6. Changements de phases (détat) des corps purs
6.1. Introduction
Le schéma ci-après donne la nomenclature des divers changements de phase (on dit aussi changements détat ou transitions de phase) entre les états solide, liquide et gazeux.
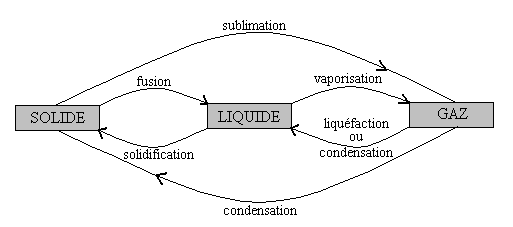
Il convient dêtre précis avec le terme condensation.
Il sagit du passage de létat dilué ou raréfié
(gazeux ou vapeur) à un état condensé (liquide ou
solide). Pour être clair, on devrait préciser, condensation
à létat liquide ou condensation à létat solide.
Souvent pour le passage à létat liquide, on emploie
lexpression liquéfaction lorsquil sagit dun gaz et lexpression
condensation lorsquil sagit dune vapeur.
6.2. Observation courante
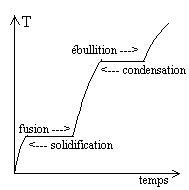 |
Lexpérience courante de transitions
de phase consiste à fournir de lénergie thermique à
un solide à pression constante.
Sa température augmente, puis il fond (la température reste constante durant la fusion). Lorsque le corps est entièrement liquide, sa température recommence à augmenter, puis il bout (sa température reste constante durant lébullition). Lorsque le corps est gazeux, sa température recommence à augmenter. Par refroidissement, on peut faire lexpérience inverse, en passant par les stades de condensation et de solidification. |
6.3. Equilibre liquide-vapeur
Etude expérimentale, courbe de saturation
Aux températures inférieures à la température critique
Tc , la compression isotherme dun gaz provoque sa liquéfaction,
cependant nous devons nous situer à des pressions supérieures
à la pression pt
du point triple où coexistent les états solide, liquide et gazeux.
Si, à partir dune pression faible où létat est
gazeux, on veut réduire le volume, il faut augmenter la pression
(un gaz est facilement compressible). A un certain niveau de pression que
nous nommons pression de vapeur saturante pV
, il apparaît une première goutte de liquide facile à
distinguer et, à partir de là, on peut réduire le
volume sans augmenter la pression. On constate que, dans lenceinte, il
y a de plus en plus de liquide. Lorsquil ny a plus que du liquide, il
faut exercer des augmentations de pression très fortes pour réduire
le volume (un liquide est, en première approximation, incompressible).
Il est possible de faire le processus expérimental inverse en
réduisant les pressions. On part de létat liquide compressé.
Lorsquon atteint la pression de vapeur saturante, il apparaît dans
l enceinte la première bulle de gaz (le liquide se met à
bouillir). Tant que le liquide est en ébullition, la pression reste
constante, le volume augmente et il y a de plus en plus de gaz. Lorsquil
ny a plus que du gaz, il faut réduire la pression pour augmenter
le volume.
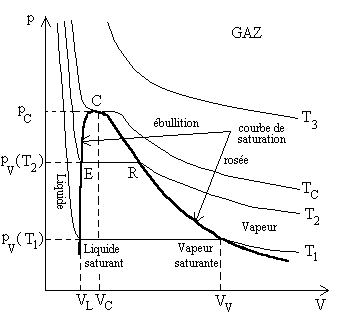 |
Les faits expérimentaux sont repré-sentés
pour différents niveaux de température tels que :
Lensemble des points E est lensemble des points où apparaît la première bulle gazeuse : ils forment la courbe débullition. Lensemble des points R est lensemble des points où apparaît la première goutte de liquide : ils forment la courbe de rosée. La courbe de saturation est formée de la courbe débullition et de la courbe de rosée. Le point C, point supérieur de la courbe de saturation est appelé le point critique. |
Ainsi pour des températures supérieures à la température
Tc du point critique, un corps ne peut exister quà létat
gazeux.
La courbe de compression isotherme, pour la température critique,
présente au point critique un point dinflexion, la tangente y est
horizontale.
En ce point, le corps pur obéit à son équation détat,
à ![]() et
à
et
à ![]() .
.
Une interprétation du point critique à partir de léquation
détat de Van der Waals ![]() donne :
donne : ![]()
La mesure des coordonnées du point critique fournit des renseignements
sur laspect microscopique de la matière.
Coordonnées du point critique de quelques corps

Vapeur sèche, vapeur saturante, liquide saturant
A une température donnée, le changement détat
liquide-gaz se produit à la pression pV
dite pression de vapeur saturante. Cette pression est inférieure
à la pression critique et supérieure à celle du point
triple.
La pression pV ne dépend que
de la température ![]() .
.
Un corps à létat gazeux devient liquide par compression
isotherme lorsquil atteint la pression de vapeur saturante. Inversement
un corps liquide devient gazeux par détente isotherme lorsquil
atteint la pression de vapeur saturante.
A des températures supérieures à la température
critique, un corps est toujours gazeux quelque soit la pression.
Rappelons que létat gazeux à des températures
inférieures à la température critique (qui devient
liquide par compression isotherme) est appelé vapeur sèche
doù le vocabulaire changement détat (ou transition de phase)
liquide-vapeur.
Dans le langage courant, on dit le gaz oxygène, le gaz azote
et la vapeur deau.
La vapeur sèche jusquà sa limite où elle atteint
la pression de vapeur saturante (elle est alors appelée vapeur
saturante) obéit aux mêmes équations détat
que les gaz, en particulier à pressions suffisamment faibles léquation
détat des gaz parfaits est adaptée.
Lorsque la pression dun liquide est celle de la vapeur saturante,
il est appelé liquide saturant.
A lintérieur de la courbe de saturation, nous avons, à
la pression de vapeur saturante, un " mélange " de liquide saturant
et de vapeur saturante que nous appelons vapeur humide.
Liquide saturant et vapeur saturante pris seuls obéissent, en un point
donné, aux équations détat du liquide ou de la vapeur
sèche dont ils sont la limite ; par contre, la vapeur humide a
un comportement tout à fait différent et léquation détat
est ![]() .
.
Vaporisation dans le vide
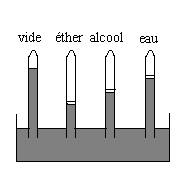 |
Nous introduisons dans les tubes des liquides
différents.
Ils se vaporisent, entièrement si les quantités introduites sont faibles, partiellement si les quantités introduites sont suffisantes (la pression est alors la pression de vapeur saturante). Les dénivellations par rapport au tube de référence sont différentes, la pression de vapeur saturante dépend du corps. A 20 °C pour léther, lalcool et leau, on obtient respectivement de pressions de vapeur saturante de 442, 44 et 17,5 mm de mercure. |
Vaporisation en atmosphère gazeuse
Contrairement à la vaporisation dans le vide qui est " instantanée
", la vaporisation dans un environnement gazeux est un phénomène
lent.
La pression dun mélange gazeux contenant plusieurs constituants dont
certains sont dans des conditions de saturation est égale à : ![]()
Le premier terme représente la somme des pressions partielles
des divers gaz et vapeurs sèches, le second terme la somme des pressions
des vapeurs saturantes.
Applications du changement détat liquide-vapeur
Létude des machines thermiques nous montrera limportance du
changement détat liquide-vapeur.
Dans ce paragraphe, nous citerons :
- la condensation de leau sur les parois froides et la migration de
lhumidité,
- les phénomènes de séchage,
- le stockage des fluides,
- lexploitation des retards aux changements de phase pour la visualisation
des trajectoires des particules, retard à la condensation dans le
cas des chambres de Wilson, retard à lébullition pour les
chambres à bulle.
6.4. Equilibre liquide-solide, équilibre solide-gaz
Les phénomènes de transitions de phase liquide-solide
et solide-gaz sont analogues à ceux que nous avons étudiés
pour la transition liquide-vapeur.
Cependant, pour ces équilibres, il ny a pas de point critique.
Le phénomène de fusion est courant, celui de sublimation
moins car la pression doit être inférieure à celle
du point triple.
Dans les conditions ordinaires, nous citerons,
- lodeur des cristaux de naphtalène,
- dans un tube à essais modérément chauffé,
on observe la coexistence de cristaux diode et de vapeurs violettes diode.
Courbes de sublimation, de fusion, de vaporisation. Point triple
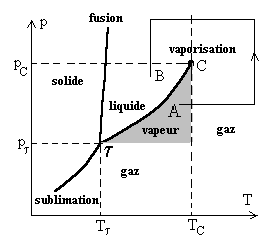 |
Dans un diagramme T, p traçons
les trois courbes déquilibre solide-gaz ou courbe de sublimation,
liquide-vapeur ou courbe de vaporisation, liquide-solide ou courbe de fusion.
Ces trois courbes se coupent nécessairement en un même point appelé point triple t . Au point triple, il y a coexistence des trois phases solide, liquide et gazeuse. Si nous faisons varier soit la pression, soit la température soient ces deux paramètres à partir du point triple, il y a disparition dune ou deux phases. |
La sublimation ne peut se produire pour des pressions supérieures
à celle du point triple.
La courbe de fusion est toujours très proche de la verticale,
généralement de pente positive. Dans le cas de leau, elle
est négative ce qui explique lexpérience amusante du " regel
de leau ".
La courbe de vaporisation est limitée par le point critique C et le point
triple t. Certaines formules
à caractère empirique plus ou moins marqué donnent de bonnes
représentations de ![]() .
.
Nous citerons, la formule de Dupré ![]() ,
celle de Rankine
,
celle de Rankine ![]() ,
celle de Duperray valable pour leau entre 100 °C et 200 °C
à savoir
,
celle de Duperray valable pour leau entre 100 °C et 200 °C
à savoir ![]() où
la pression est en atmosphères et la température en Celsius.
où
la pression est en atmosphères et la température en Celsius.
Enfin, au risque de nous répéter, nous croyons important
dinsister :
- les phénomènes de transitions de phase sont nettement
marqués visuellement (formation de bulles dans un liquide ou dun
solide, dun liquide à la surface dun solide ou dans une vapeur)
et accompagnés de " discontinuités " des propriétés
physiques pour des températures inférieures à la température
critique,
- si nous considérons la transformation AB représentée
sur la figure ci-dessus, nous passons par les états vapeur, gaz
et liquide sans phénomène visuel fortement marqué.
Il y a continuité de létat gazeux et de létat
liquide, ces deux phases appartiennent à un même état
appelé fluide et diffèrent par une plus ou moins grande densité
des molécules.
Il ne faut donc pas sétonner que les liquides obéissent
à des équations détat de même type que les
gaz.
| La figure ci-contre représente les transitions de phase solide, liquide et gaz dans un diagramme p, V . | 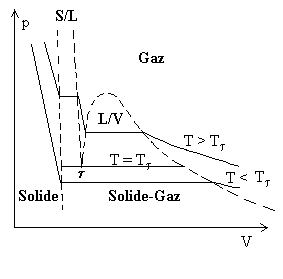 |
6.5. Remarque
Les changements de phase des corps purs saccompagnent déchanges
dénergie considérables.
Cet aspect est abordé dans létude des transferts dénergie.