Chapitre 4 : Structure de la matière
Plan
1. Introduction : gaz, liquide, solide
2. L’hypothèse atomique (ou moléculaire)
3. Les deux états de la matière : l'état
désordonné et l'état ordonné
3.1. Les états désordonnés
3.1.1. Le Gaz
3.1.1.1. Agitation au niveau microscopique
3.1.1.2. Interprétation microscopique de la pression
3.1.1.3. Mélange idéal de gaz parfaits
3.1.1.4. Facteur de Boltzmann. Raréfaction de
l'air
3.1.1.5. Température cinétique. Equipartition
de l'énergie. Energie d’un gaz parfait
3.1.2. Le liquide
3.1.3. Le solide amorphe
3.2. L'état ordonné : le solide cristallisé
4. Changements de phases (d’état) des corps purs
4.1. Introduction
4.2. Observation courante
4.3. Equilibre liquide-vapeur
4.4. Equilibre liquide-solide, équilibre solide-gaz
4.5. Remarque
Les disciplines scientifiques tels que la Physique, la Chimie,
les Sciences de la Vie, de la Terre, de l'Univers, de l'Environnement reposent
sur des modèles, des représentations de la matière.
Dans l'enseignement secondaire, les premières leçons de Chimie
introduisent des notions d’atomistique, de liaisons et d’édifices entre
les atomes.
Dans notre vie courante et dans nos premiers apprentissages scientifiques, nous
disons de la matière qu'elle est solide, liquide, gazeuse, vapeur, fluide.
Nous savons "parfaitement" faire passer la matière d'un état à
un autre : transformer un solide en liquide (exemple : faire fondre un glaçon),
transformer un liquide en gaz (à moins que ce ne soit en vapeur ; exemple
: faire bouillir de l'eau).
Jusqu'à présent dans ce cours, pour
nous représenter la matière, nous avons utilisé notre sens
commun et avons pu, sans trop de difficulté de compréhension entre
nous, donner un contenu scientifique à des notions aussi tangibles que
pression ou température.
Et pourtant,
cette matière donc sommes-nous capables, dans l'état actuel de nos connaissances, d'en parler avec précision ?
1. Introduction : gaz, liquide, solide
Communément, on classe les milieux matériels
en solides et fluides
Ces deux notions, comme beaucoup de notions premières, ne sont pas
simples à définir, la difficulté augmente si l'on souhaite,
comme tout un chacun, distinguer les liquides et les gaz à l'intérieur
de la famille fluide.
Il n’est jamais inutile d’ouvrir un dictionnaire et de chercher le sens qui
peut être donné aux mots.
Ouvrons (sans en faire publicité) le "Petit Robert" et regardons les définitions à usage courant.
Fluide : "Qui n'est ni solide, ni épais, coule aisément". Mais l'huile, par exemple, c'est épais ou non ? Allons voir à solide.
Solide : "Qui a de la consistance, qui n'est pas liquide, tout en pouvant être plus ou moins mou". Qui n'est pas liquide ? Allons voir à liquide.
Liquide : "Tout corps qui coule ou tend à couler". Peut-on dire que le sable sec qui coule entre les doigts est un liquide ?
Les définitions à usage courant semblent bien floues, aussi dans le même dictionnaire nous considérons les définitions scientifiques.
Fluide : "Tout corps qui se laisse déformer sous l'action de forces minimes; tout corps qui épouse la forme de son contenant"
Solide : "Se dit d'un corps dans lequel les molécules sont très rapprochées les unes des autres et vibrent avec une très faible amplitude autour de leur position d'équilibre; qui a de la cohésion, garde une forme relativement constante lorsqu'il n'est pas soumis à des forces extérieures"
Liquide : "Tout corps à l'état fluide, pratiquement incompressible, formé de corpuscules (ions ou molécules) soumis à de faibles attractions"
Gaz : "Tout corps qui se présente à l'état de fluide expansible et compressible (état gazeux) dans les conditions de température et de pression normales"
Cette lecture permet de dégager quelques pistes :
Sans doute, avons nous l’impression d’en savoir plus, mais pour progresser on devra " prendre un microscope " c'est à dire faire intervenir une conception microscopique de la matière.
2. L’hypothèse atomique (ou moléculaire)
L’hypothèse est banale (universellement admise). Elle
était déjà émise par les philosophes de la Grèce
antique. Par contre les premières preuves expérimentales - Dalton,
Proust, Lavoisier au 19ème siècle
- sont relativement récentes.
Si, dans un cataclysme, toute notre connaissance scientifique devait être détruite et qu’une seule phrase passe aux générations futures, quelle affirmation contiendrait le maximum d’information dans le minimum de mots ? N’est ce pas l’hypothèse atomique à savoir " toutes les choses sont faites d’atomes (de molécules), petites particules en agitation incessante, s’attirant mutuellement à petite distance les unes des autres et se repoussant si on cherche à trop les rapprocher ". Dans cette seule phrase vous verrez qu’il y a une énorme quantité d’informations sur le monde, si on lui applique un petit peu d’imagination et de réflexion. The Feynman Lectures on Physics par Richard P. Feynman, Robert B. Leighton et Mattews Sands.
Sans doute, cette phrase de Richard P. Feynman, prix Nobel de Physique en 1965, est suffisante pour aborder un premier cours de Thermodynamique à condition de la compléter par :
Dans un cadre limité à l'enseignement de la Physique, cette phrase est très insuffisante pour comprendre le comportement de la matière soumise à un champ électrique et/ou magnétique, pour aborder l’interaction onde-matière c’est à dire une physique dite " moderne " celle qui ne s’est construite qu’à partir du début du 20ème siècle. Le lecteur intéressé pourra compléter ces connaissances par l'annexe "Les particules de base de la matière" et par l'étude de cours ou de documents de type "Atomistique et Structure"
La compressibilité des gaz, la presque incompressibilité des liquides et des solides permet de dire que les atomes d’un liquide ou d’un solide sont " entassés " les uns sur les autres alors que ceux d’un gaz sont éloignés les uns des autres. Ceci est confirmé par la masse volumique qui est faible (peu d’atomes par unité de volume) pour les gaz et importante pour les liquides et les solides, par le fait que nous nous sentons " léger " (forte poussée d’Archimède) dans l’eau et " lourd " dans l’air, que nous avons du mal à marcher dans l’eau, …
L’état gazeux est un état dilué alors que liquides et solides font partie des états condensés.
3. Les deux états de la matière : l'état désordonné et l'état ordonné
Le sens commun nous fait distinguer trois états de la
matière : solide, liquide et gazeux ... sans que nous soyons capables
d'en faire les distinguo de manière rigoureuse.
Le modèle atomique de la matière conduit à une séparation
des états physiques en deux classes dont le mérite est une coupure
plus franche : le première classe est dite à structure désordonnée,
la deuxième à structure ordonnée.
3.1.1.1. Agitation au niveau microscopique
Les modèles ne sont jamais statiques (ce qui n'est pas
en contradiction avec " globalement au repos " au niveau
macroscopique).
C'est particulièrement évident pour un gaz où la matière
est diluée c’est à dire où les particules sont " libres
" de leur déplacement. En effet, si elles n'étaient pas en
mouvement, du fait du champ de pesanteur, elles ne pourraient qu'être
entassées à partir du fond du récipient qui les contient
ou à partir du sol ce qui ne peut manifestement être le cas pour
une substance diluée.
Ainsi dans les gaz, s’introduit la notion de mouvement sans cesse des particules,
dans toutes les directions au gré des chocs, que l'on appelle agitation.
Les particules seront attirées vers les altitudes les plus faibles mais
sans pouvoir s'y entasser.
Dans un gaz, les interactions entre particules sont faibles (matière
diluée, particules en moyenne éloignées les unes des autres),
les mouvements de celles-ci sont assez libres, il règne un désordre,
que l'on qualifie de parfait si on néglige toute interaction entre
particules.
3.1.1.2. Interprétation microscopique de la pression
Une conséquence de l'agitation est une visualisation
locale (au niveau microscopique) de la pression.
La pression est la force moyenne par unité de surface due aux particules
venant frapper une paroi.
La pression augmente :
- avec le nombre de chocs contre la paroi donc, dans une enceinte de volume
V, avec le nombre N de particules (soit, en fait, avec N/V),
- avec l'agitation des particules puisque la quantité de mouvement transférée
de la particule à la paroi sera d'autant plus importante.
Notons l'accord entre le caractère non directionnel de la pression et le fait que le mouvement des particules ait lieu dans toutes les directions.
De même pour que la pression tende vers une valeur nulle (comportement limite de tous les gaz réels appelé gaz parfait), il convient que le nombre de chocs soit faible, c’est à dire que la densité des particules soit faible (gaz raréfié), c’est à dire qu’en moyenne elles soient éloignées les unes des autres, donc que les interactions entre les particules soient négligeables. La probabilité pour que deux particules entrent en collision est extrêmement faibles, chaque particule rebondit d'une paroi à une autre.
3.1.1.3. Mélange idéal de gaz parfaits
Dans une enceinte de volume V, nous plaçons différentes substances chimiques en phases gazeuses sans possibilité de réactions chimiques entre elles. A cause de l'agitation les substances se mélangent, le mélange est dit idéal.
Si p la pression du mélange est suffisamment
faible, l’expérience montre que :
![]() est le
nombre de moles du gaz i et n le nombre total de moles gazeuses sans
distinction de la substance gazeuse.
est le
nombre de moles du gaz i et n le nombre total de moles gazeuses sans
distinction de la substance gazeuse.
Le mélange se comporte comme un gaz parfait.
Si le gaz i était seul dans l’enceinte, il se comporterait comme
un gaz parfait et sa pression ![]() serait donnée par la relation
serait donnée par la relation ![]() .
.
On remarquera que la pression p dans le mélange est liée
aux pressions ![]() par
la relation
par
la relation ![]() .
.
![]() est appelée
pression partielle du gaz i dans le mélange. La pression du mélange
est la somme des pressions partielles.
est appelée
pression partielle du gaz i dans le mélange. La pression du mélange
est la somme des pressions partielles.
On appelle fraction molaire du gaz i dans le mélange, la quantité
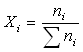 .
.
On déduit que ![]() .
.
La quantité ![]() représente la masse totale du mélange si
représente la masse totale du mélange si ![]() est la masse molaire du gaz i.
est la masse molaire du gaz i.
On définit la " masse molaire du mélange "
par 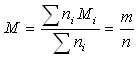 .
.
Autre écriture de l’équation d’état des gaz parfaits
pV = nRT s’écrit aussi pV = (m/M) RT = mrT
où r = R/M.
Si N est le nombre de molécules du gaz, N = nN
où N est le nombre d’Avogadro.
PV = nRT ![]() pV = NkT où
k = R/N = 1,38 10-23
est la constante de Boltzmann.
pV = NkT où
k = R/N = 1,38 10-23
est la constante de Boltzmann.
L’équation d’état des gaz parfaits s’écrit encore ![]() en introduisant
en introduisant ![]() la densité volumique molaire et
la densité volumique molaire et ![]() la densité volumique des molécules.
la densité volumique des molécules.
Remarque : localement la pression est proportionnelle à la densité
des molécules
Masse volumique et densité des gaz parfaits
Soit m = m/V la masse volumique.
On obtient ![]() (autre
écriture de l’équation d’état des gaz parfaits).
(autre
écriture de l’équation d’état des gaz parfaits).
On appelle densité d’un gaz le rapport de la masse d’un certain volume
de gaz sur la masse du même volume d’air pris dans les mêmes conditions
de pression et de température.
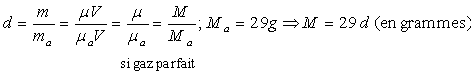
3.1.1.4. Facteur de Boltzmann. Raréfaction de l'air
Nous traitons là un exemple de statique des fluides
compressibles.
Nous cherchons à savoir comment varie la pression p avec l’altitude
z. Nous formulons l’hypothèse justifiée que l’air
est un mélange idéal de gaz se comportant comme un gaz parfait
(nous savons que la pression est de l'ordre de 1 atmosphère au sol et
diminue avec l'altitude) et celle fausse dans la troposphère (altitude
comprise entre 0 et 20 km) que la température est la
même quelque soit l’altitude.
Soient ![]() la température
et
la température
et ![]() la masse molaire
de l’air.
la masse molaire
de l’air.
Nous utilisons le principe de l’hydrostatique dp = -mg
dz et l’équation d’état des gaz parfaits.
![]()
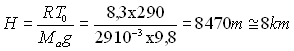
A température uniforme, la pression est proportionnelle
à la masse volumique ainsi qu’à la densité volumique molaire.
On obtient :
![]()
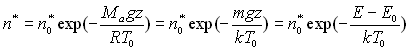
![]()
où ![]() représente
la masse " moyenne " d’une molécule d’air et
représente
la masse " moyenne " d’une molécule d’air et ![]() est
l’énergie potentielle dans le champ de pesanteur terrestre de la " molécule
d’air " à l’altitude z.
est
l’énergie potentielle dans le champ de pesanteur terrestre de la " molécule
d’air " à l’altitude z.
Cette application nous explique le phénomène de raréfaction de l’air avec l’altitude, à savoir la densité moléculaire diminue avec l’altitude suivant une fonction dépendant de l’énergie de la molécule à cette altitude et de la température.
La fonction 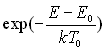 appelée facteur de Boltzmann est d’une importance considérable
en Physique statistique.
appelée facteur de Boltzmann est d’une importance considérable
en Physique statistique.
Dans l'exemple considéré, elle s’interprète
de la manière suivante :
le rapport ![]() est
la probabilité de présence d'une molécule à l'altitude
z. Si, pour une altitude, ce rapport est égal à 1/2, ceci veut
dire qu'il y a deux fois moins de molécules à cette altitude qu'à
l'altitude de référence ou, dit différemment, que la probabilité
de présence d'une molécule à l'altitude z est 50% de celle
de la trouver à l'altitude de référence.
est
la probabilité de présence d'une molécule à l'altitude
z. Si, pour une altitude, ce rapport est égal à 1/2, ceci veut
dire qu'il y a deux fois moins de molécules à cette altitude qu'à
l'altitude de référence ou, dit différemment, que la probabilité
de présence d'une molécule à l'altitude z est 50% de celle
de la trouver à l'altitude de référence.
3.1.1.5. Température cinétique. Equipartition de l'énergie. Energie d’un gaz parfait
La distribution des vitesses des centres de masse des molécules
d’un gaz parfait en équilibre à température T vérifie
le facteur de Boltzmann c'est à dire que le nombre de particules dont
le centre de masse a une composante de vitesse ![]() est proportionnelle à
est proportionnelle à 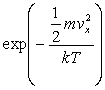
Ce résultat est connu sous le nom de loi distribution
des vitesses de Maxwell-Boltzmann et entre dans le cadre de la théorie
cinétique du gaz parfait (voir Annexe)
Résultat des travaux de Maxwell et de Boltzmann à la fin du 19ème
siècle, elle a été validée expérimentale
par les expériences sur les " jets monoatomiques ".
Il convient de citer la première en 1947 de Estermann, Simpson et Stern
et celle très convaincante réalisée en 1959 par Marcus
et McFee.
Nous retiendrons deux résultats fondamentaux :
Suivant ce résultat, la température est une mesure de l'agitation (on parle d'agitation thermique ou de température cinétique).
Ce résultat est général en Physique et constitue le théorème d'équipartition de l'énergie.
Remarque : en remplaçant dans l’équation
d’état du gaz parfait, on obtient ![]() relation tout à fait cohérente avec l’interprétation microscopique
de la pression.
relation tout à fait cohérente avec l’interprétation microscopique
de la pression.
Energie interne d’un gaz parfait
L’énergie interne d’un gaz parfait est constituée
des énergies cinétiques des molécules dans leur mouvement
et de la somme (que nous notons ![]() des énergies internes à chaque molécule. Les énergies
d’interaction entre molécules sont négligeables (état raréfié).
des énergies internes à chaque molécule. Les énergies
d’interaction entre molécules sont négligeables (état raréfié).
Pour un gaz monoatomique de N molécules
où l’énergie cinétique des molécules est l’énergie
de translation du centre de masse, on obtient : ![]() qui correspond à la décomposition d’un mouvement de translation
quelconque en trois mouvements de translation indépendants (ð
3 degrés de liberté).
qui correspond à la décomposition d’un mouvement de translation
quelconque en trois mouvements de translation indépendants (ð
3 degrés de liberté).
En Calorimétrie, on accède par la mesure
à la quantité ![]() appelé capacité calorifique à volume constant.
appelé capacité calorifique à volume constant.
Le graphique ci-après montre, pour une mole de gaz monoatomique à
pression faible, des résultats tout à fait conformes à
la théorie (sauf aux très basses températures).
Pour les gaz polyatomiques, les résultats expérimentaux sont plus
complexes : pour les diatomiques, la capacité calorifique molaire
à volume constant ![]() est nulle à 0 K, elle atteint la valeur
est nulle à 0 K, elle atteint la valeur ![]() à partir de 2-3 K, puis
à partir de 2-3 K, puis ![]() à partir de 150 K,
à partir de 150 K, ![]() à très hautes températures.
à très hautes températures.
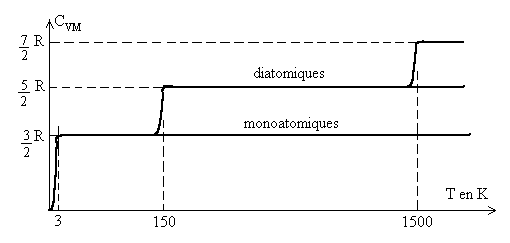
Pour expliquer ces augmentations des valeurs de la capacité
calorifique, il faut ajouter des sources d’énergie : énergie
de rotation des molécules correspondant à deux degrés
de liberté (deux rotations indépendantes) et l’énergie
de vibration des atomes constituant les molécules (deux degrés
de liberté correspondant à de l’énergie cinétique
et à de l’énergie potentielle). On trouve donc une énergie
supérieure à celle trouvée pour un gaz monoatomique.
Pour des molécules d’atomicité supérieure, les résultats
expérimentaux se compliquent mais l’interprétation en termes d’énergie
de translation, de rotation et de vibration demeure.
Remarque : on ne peut comprendre les évolutions de ![]() avec la température sans notion de mécanique quantique et de quantum
d’énergie.
avec la température sans notion de mécanique quantique et de quantum
d’énergie.
Agitation thermique dans les liquides
La notion d'agitation est moins évidente puisqu'un liquide
est entassé au fond du récipient. Remarquons cependant que si
nous mettons deux liquides miscibles dans un récipient, spontanément
ils diffusent c'est à dire ils se mélangent de manière
à former un liquide homogène.
Donc pour le liquide, le modèle statique au niveau microscopique n'est
pas possible. Les molécules de liquide sont soumises à l'agitation
thermique, leurs mouvements ne sont pas libres comme dans un gaz car elles sont
proches les unes des autres (les liquides ont des masses volumiques importantes
et constituent des milieux denses, condensés)
Un liquide est un état moins désordonné qu'un gaz.
Beaucoup de molécules encastrées entre leurs
voisines ne peuvent que vibrer. Il peut arriver qu'une molécule puisse
s'échapper et échanger sa position avec une autre : c'est l'origine
du phénomène de diffusion.
Pour les liquides, il n’y a pas de résultats (ni de théories)
simples
La viscosité des liquides
Quand un liquide n'est pas en équilibre au fond d'un récipient, il coule. Les molécules glissent les unes sur les autres, elles sont maintenues en une masse compacte de volume constant par les forces de cohésion interne qui n'empêchent pas la forme extérieure du liquide de se modifier. Si le glissement est facile, le liquide est dit fluide et la viscosité est faible. Si la viscosité est très élevée, le liquide ressemble à un solide mou. La viscosité est très sensible à la température.
Le cas typique est celui du verre qui, à température
ordinaire, a l'apparence d'un solide très dur et indéformable.
Lorsqu'on augmente la température, il fond sans qu'il y ait discontinuité
dans les propriétés physiques (transition progressive), c'est
à dire sans qu'il y ait de changement dans la structure atomique.
C'est un cas rare de solide appelé solide amorphe ou vitreux
où l'état solide est en fait un état liquide (donc une
structure désordonnée) de viscosité très élevée.
3.2. L'état ordonné : le solide cristallisé
Pour l'immense majorité des corps purs, la transition
solide-liquide est discontinue et se produit à température fixe
pour une pression déterminée : suivant le sens, elle est appelée
fusion ou solidification.
Il existe aussi une transition discontinue solide-gaz : suivant le sens, c'est
la sublimation ou la condensation.
Les solides (à l’exception des solides amorphes) ont des structures ordonnées. On parle de solide cristallisé avec des maillages périodiques, des " empilements " réguliers. C’est le domaine de la cristallographie.
On distingue quatre types de solides cristallisés.
L’énergie d’un solide est constituée de la somme
des énergies de vibration (autour de leur position d’équilibre)
des particules de base (ions, atomes ou molécules) et de la somme des
énergies internes des particules de base.
Si la particule de base est un atome ou un ion simple, on obtient, si la température
est suffisante, ![]() .
.
Ce résultat est connu sous le nom de loi de Dulong et Petit
Remarque : entre ordre et désordre
Dans un souci de clarté, nous avons distingué
l'état ordonné et l'état désordonné.
En fait, la situation est parfois plus complexe et on parle de structures mal
cristallisées ou douées d'un ordre cristallin partiel
: dans cette catégorie, on range les hauts polymères (plastiques),
beaucoup de substances essentielles à la constitution de nos organes
et les cristaux liquides.
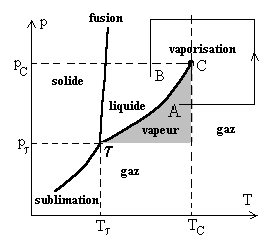
4. Changements de phases (d’état) des corps purs
Le schéma ci-après donne la nomenclature des divers changements de phase (on dit aussi changements d’état ou transitions de phase) entre les états solide, liquide et gazeux.
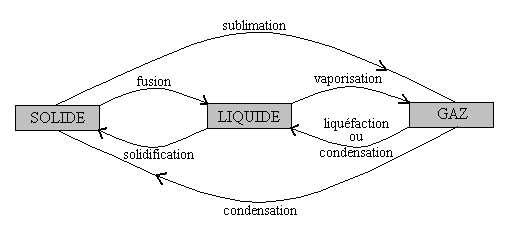
Il convient d’être précis avec le terme condensation.
Il s’agit du passage de l’état dilué ou raréfié
(gazeux ou vapeur) à un état condensé (liquide ou solide).
Pour être clair, on devrait préciser, condensation à l’état
liquide ou condensation à l’état solide.
Souvent pour le passage à l’état liquide, on emploie l’expression
liquéfaction lorsqu’il s’agit d’un gaz et l’expression condensation lorsqu’il
s’agit d’une vapeur.
|
|
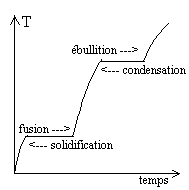
L’expérience courante de transitions de phase
consiste à fournir de l’énergie thermique à un solide
à pression constante. |
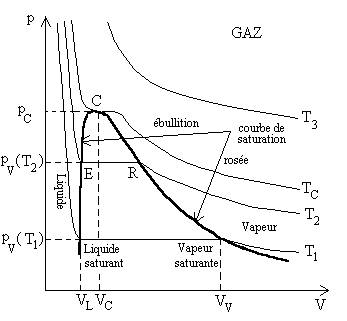
Etude expérimentale, courbe de saturation
Aux températures inférieures à la température
critique TC , la compression isotherme
d’un gaz provoque sa liquéfaction, cependant nous devons nous situer
à des pressions supérieures à la pression pt
du point triple où coexistent les états solide, liquide et gazeux.
Si, à partir d’une pression faible où l’état est gazeux,
on veut réduire le volume, il faut augmenter la pression (un gaz est
facilement compressible). A un certain niveau de pression que nous nommons pression
de vapeur saturante pV , il apparaît
une première goutte de liquide facile à distinguer et, à
partir de là, on peut réduire le volume sans augmenter la pression.
On constate que, dans l’enceinte, il y a de plus en plus de liquide. Lorsqu’il
n’y a plus que du liquide, il faut exercer des augmentations de pression très
fortes pour réduire le volume (un liquide est, en première approximation,
incompressible).
Il est possible de faire le processus expérimental inverse en réduisant
les pressions. On part de l’état liquide compressé. Lorsqu’on
atteint la pression de vapeur saturante, il apparaît dans l ’enceinte
la première bulle de gaz (le liquide se met à bouillir). Tant
que le liquide est en ébullition, la pression reste constante, le volume
augmente et il y a de plus en plus de gaz. Lorsqu’il n’y a plus que du gaz,
il faut réduire la pression pour augmenter le volume.
|
|
Les faits expérimentaux sont repré-sentés
pour différents niveaux de température tels que : |
Ainsi pour des températures supérieures à
la température TC du point critique,
un corps ne peut exister qu’à l’état gazeux.
La courbe de compression isotherme, pour la température critique,
présente au point critique un point d’inflexion, la tangente y est horizontale.
En ce point, le corps pur obéit à son équation
d’état, à ![]() et à
et à ![]() .
.
Une interprétation du point critique à partir de l’équation
d’état de Van der Waals ![]() donne :
donne : ![]()
La mesure
des coordonnées du point critique fournit des renseignements sur l’aspect
microscopique de la matière.
Coordonnées du point critique de quelques corps
|
Corps |
TC en K |
pC en atm |
Corps |
TC en K |
pC en atm |
||||
|
Air |
132,4 |
36 |
Méthane |
190,2 |
45 |
||||
|
CO2 |
304,1 |
71 |
O2 |
154,2 |
49 |
||||
|
N2 |
125,9 |
32 |
Propane |
370,6 |
39 |
||||
|
Butane |
426,3 |
35 |
H2 |
32,1 |
19 |
||||
|
He |
5,2 |
2,3 |
H2O |
647,3 |
218 |
||||
|
Isobutane |
406,8 |
36 |
|||||||
Vapeur sèche, vapeur saturante, liquide saturant
A une température donnée, le changement d’état liquide-gaz se produit à la pression pV dite pression de vapeur saturante. Cette pression est inférieure à la pression critique et supérieure à celle du point triple.
La pression pV ne dépend
que de la température ![]() .
.
Un corps à l’état gazeux devient liquide par compression isotherme lorsqu’il atteint la pression de vapeur saturante. Inversement un corps liquide devient gazeux par détente isotherme lorsqu’il atteint la pression de vapeur saturante.
A des températures supérieures à la température critique, un corps est toujours gazeux quelque soit la pression.
Rappelons que l’état gazeux à des températures
inférieures à la température critique (qui devient liquide
par compression isotherme) est appelé vapeur sèche d’où
le vocabulaire changement d’état (ou transition de phase) liquide-vapeur.
Dans le langage courant, on dit le gaz oxygène, le gaz azote et la
vapeur d’eau.
La vapeur sèche jusqu’à sa limite où elle atteint la pression
de vapeur saturante (elle est alors appelée vapeur saturante)
obéit aux mêmes équations d’état que les gaz, en
particulier à pressions suffisamment faibles l’équation d’état
des gaz parfaits est adaptée.
Lorsque la pression d’un liquide est celle de la vapeur saturante, il est appelé
liquide saturant.
A l’intérieur de la courbe de saturation, nous avons, à la pression
de vapeur saturante, un " mélange " de liquide saturant
et de vapeur saturante que nous appelons vapeur humide.
Liquide saturant
et vapeur saturante pris seuls obéissent, en un point donné, aux
équations d’état du liquide ou de la vapeur sèche dont
ils sont la limite ; par contre, la vapeur humide a un comportement tout
à fait différent et l’équation d’état est ![]() .
.
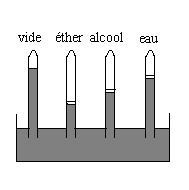
Vaporisation dans le vide
|
|
Nous introduisons dans les tubes des liquides différents. |
Vaporisation en atmosphère gazeuse
Contrairement à la vaporisation dans le vide qui
est " instantanée ", la vaporisation dans un environnement
gazeux est un phénomène lent.
La pression d’un mélange gazeux contenant plusieurs constituants
dont certains sont dans des conditions de saturation est égale à
:
![]()
Le premier terme
représente la somme des pressions partielles des divers gaz et vapeurs
sèches, le second terme la somme des pressions des vapeurs saturantes.
Applications du changement d’état liquide-vapeur
L’étude des machines thermiques nous montrera l’importance du changement d’état liquide-vapeur.
Dans ce paragraphe, nous citerons :
4.4. Equilibre liquide-solide, équilibre solide-gaz
Les phénomènes de transitions de phase liquide-solide
et solide-gaz sont analogues à ceux que nous avons étudiés
pour la transition liquide-vapeur.
Cependant, pour ces équilibres, il n’y a pas de point critique.
Le phénomène de fusion est courant, celui de sublimation moins
car la pression doit être inférieure à celle du point triple.
Dans les conditions ordinaires, nous citerons,
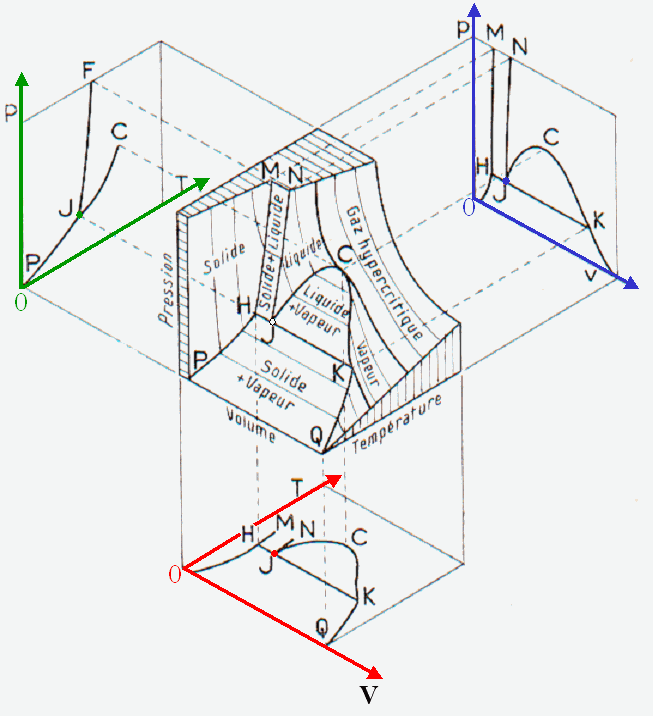
Courbes de sublimation, de fusion, de vaporisation. Point triple
|
|
Dans un diagramme T, p traçons les trois
courbes d’équilibre solide-gaz ou courbe de sublimation, liquide-vapeur
ou courbe de vaporisation, liquide-solide ou courbe de fusion. |
La sublimation ne peut se produire pour des pressions supérieures
à celle du point triple.
La courbe de fusion est toujours très proche de la verticale, généralement
de pente positive. Dans le cas de l’eau, elle est négative ce qui explique
l’expérience amusante du " regel de l’eau ".
La courbe de vaporisation est limitée par le point critique C et le point
triple t. Certaines formules à caractère
empirique plus ou moins marqué donnent de bonnes représentations
de ![]() .
.
Nous citerons, la formule de Dupré ![]() ,
celle de Rankine
,
celle de Rankine ![]() ,
celle de Duperray valable pour l’eau entre 100 °C et 200 °C à
savoir
,
celle de Duperray valable pour l’eau entre 100 °C et 200 °C à
savoir ![]() où la pression est
en atmosphères et la température en Celsius.
où la pression est
en atmosphères et la température en Celsius.
Enfin, au risque de nous répéter, nous croyons important d’insister :
Il y a continuité de l’état gazeux et de l’état liquide, ces deux phases appartiennent à un même état appelé fluide et diffèrent par une plus ou moins grande densité des molécules.
Il ne faut donc pas s’étonner que les liquides obéissent à des équations d’état de même type que les gaz.
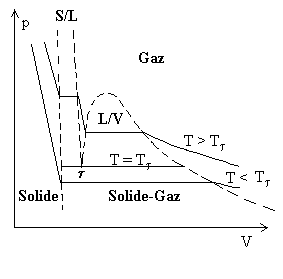
|
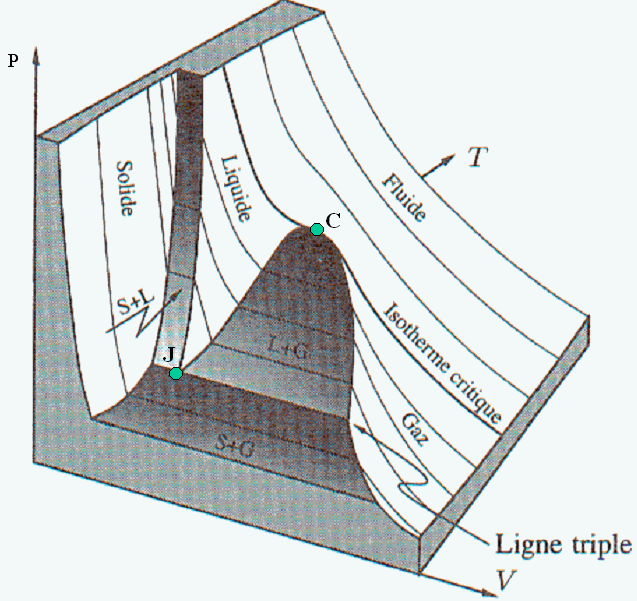
La figure ci-contre représente les transitions de phase solide, liquide et gaz dans un diagramme p, V . |
|
Les changements de phase des corps pur s’accompagnent d’échanges d’énergie considérables.
Cet aspect sera abordé dans un chapitre ultérieur
sur l'étude énergétique des changements de phases.
Représentation dans un espace à trois dimensions ( p, V, T ) des trois états de la matière d’un corps pur