
DCO SCF Jean-Pierre Sauvage prize awarded to Simon Pascal
The Division of Organic Chemistry of the French Chemical Society has awarded the “Jean-Pierre Sauvage” Young Researcher prize to Simon Pascal. His research stands out in the field of synthesis of “exotic” organic dyes and the modulation of their optical properties, particularly in the near-infrared range, opening up new perspectives in areas such as bio-imaging...

CNRS bronze medal : Morgane Vacher
As a reminder, Morgane VACHER is pursuing a very promising career in the ModES team following her recruitment in 2019. She was awarded an ERC starting grant in 2022 and has co-authored more than 48 international publications and 6 book chapters. Morgane has also been invited to speak at some twenty leading international conferences. This...
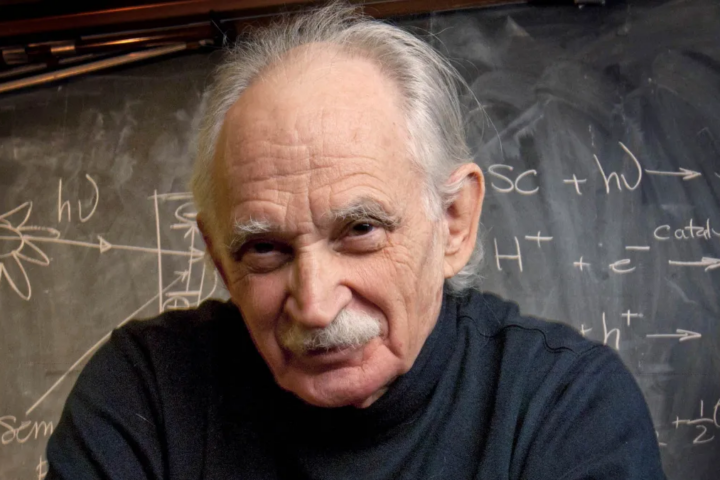
Tribute : Allen Joseph Bard, the pioneer and father of electrochemistry
Allen Joseph Bard, the pioneer and father of electrochemistry, passed away on February 11, 2024 in Austin, Texas. He was 90 years old and had a 63-year career at The University of Texas at Austin. For most of his students, postdoc and co-workers, he was the greatest electrochemist of several generations. He will be highly...




